
Running StringTie
The generic command line for the default usage has this format::stringtie [-o <output.gtf>] [other_options] <read_alignments.bam>
The main input of the program (<read_alignments.bam>) must be a SAM, BAM or CRAM file with RNA-Seq read alignments sorted by their genomic location (for example the accepted_hits.bam file produced by TopHat or the output of HISAT2 after sorting and converting it using samtools as explained below).
The main output is a GTF file containing the structural definitions of the transcripts assembled by StringTie from the read alignment data. The name of the output file should be specified with the -o option. If this option is not used the output GTF records with the assembled transcripts will be printed to the standard output (and can be captured into a file using the > output redirect operator). Note:if the --mix option is used, StringTie expects two alignment files to be given as positional parameters, in a specific order: the short read alignments must be the first file given while the long read alignments must be the second input file. Both alignment files must be sorted by genomic location.
stringtie [-o <output.gtf>] --mix [other_options] <short_read_alns.bam> <long_read_alns.bam>
Note that the command line parser in StringTie allows arbitrary order and mixing of the positional parameters with the other options of the program, so the input alignment files can also precede or be given in between the other options -- the following command line is equivalent to the one above:
stringtie <short_read_alns.bam> <long_read_alns.bam> --mix [other_options] [-o <output.gtf>]
StringTie options
The following optional parameters can be specified when running stringtie:
| -h/--help | Prints help message and exits. | ||||||||||||||||||
| --version | Prints version and exits. | ||||||||||||||||||
| -L | long reads processing mode; also enforces -s 1.5 -g 0 (default:false) | ||||||||||||||||||
| --mix | mixed reads processing mode; both short and long read data alignments are expected (long read alignments must be given as the 2nd BAM/CRAM input file) | ||||||||||||||||||
| -e | this option directs StringTie to operate in expression estimation mode; this limits the processing of read alignments to estimating the coverage of the transcripts given with the -G option (hence this option requires -G). | ||||||||||||||||||
| -v | Turns on verbose mode, printing bundle processing details. | ||||||||||||||||||
| -o [<path/>]<out.gtf> | Sets the name of the output GTF file where StringTie will write the assembled transcripts. This can be specified as a full path, in which case directories will be created as needed. By default StringTie writes the GTF at standard output. | ||||||||||||||||||
| -p <int> | Specify the number of processing threads (CPUs) to use for transcript assembly. The default is 1. | ||||||||||||||||||
| -G <ref_ann.gff> | Use a reference annotation file (in GTF or GFF3 format) to guide the assembly process. The output will include expressed reference transcripts as well as any novel transcripts that are assembled. This option is required by options -B, -b, -e, -C (see below). | ||||||||||||||||||
| --rf | Assumes a stranded library fr-firststrand. | ||||||||||||||||||
| --fr | Assumes a stranded library fr-secondstrand. | ||||||||||||||||||
| --ptf <f_tab> |
Loads a list of point-features from a text feature file
<f_tab> to guide the transcriptome assembly.
Accepted point features are transcription start sites (TSS) and
polyadenylation sites (CPAS). There are four tab-delimited columns in the feature
file. The first three define the location of the point feature on
the cromosome (sequence name, coordinate and strand), and the last
is the type of the feature (TSS or CPAS). For instance:
| ||||||||||||||||||
| -l <label> | Sets <label> as the prefix for the name of the output transcripts. Default: STRG | ||||||||||||||||||
| -f <0.0-1.0> | Sets the minimum isoform abundance of the predicted transcripts as a fraction of the most abundant transcript assembled at a given locus. Lower abundance transcripts are often artifacts of incompletely spliced precursors of processed transcripts. Default: 0.01 | ||||||||||||||||||
| -m <int> | Sets the minimum length allowed for the predicted transcripts. Default: 200 | ||||||||||||||||||
| -A <gene_abund.tab> | Gene abundances will be reported (tab delimited format) in the output file with the given name. | ||||||||||||||||||
| -C <cov_refs.gtf> | StringTie outputs a file with the given name with all transcripts in the provided reference file that are fully covered by reads (requires -G). | ||||||||||||||||||
| -a <int> | Junctions that don't have spliced reads that align across them with at least this amount of bases on both sides are filtered out. Default: 10 | ||||||||||||||||||
| -j <float> | There should be at least this many spliced reads that align across a junction (i.e. junction coverage). This number can be fractional, since some reads align in more than one place. A read that aligns in n places will contribute 1/n to the junction coverage. Default: 1 | ||||||||||||||||||
| -t | This parameter disables trimming at the ends of the assembled transcripts. By default StringTie adjusts the predicted transcript's start and/or stop coordinates based on sudden drops in coverage of the assembled transcript. | ||||||||||||||||||
| -c <float> | Sets the minimum read coverage allowed for the predicted transcripts. A transcript with a lower coverage than this value is not shown in the output. Default: 1 | ||||||||||||||||||
| -s <float> | Sets the minimum read coverage allowed for single-exon transcripts. Default: 4.75 | ||||||||||||||||||
| --conservative | Assembles transcripts in a conservative mode. Same as -t -c 1.5 -f 0.05 | ||||||||||||||||||
| -g <int> | Minimum locus gap separation value. Reads that are mapped closer than this distance are merged together in the same processing bundle. Default: 50 (bp) | ||||||||||||||||||
| -B |
This switch enables the output of Ballgown input table files (*.ctab) containing coverage data for the reference transcripts
given with the -G option. (See the Ballgown documentation for a description
of these files.) With this option StringTie can be used as a direct replacement of the tablemaker program included
with the Ballgown distribution.
If the option -o is given as a full path to the output transcript file, StringTie will write the *.ctab files in the same directory as the output GTF. | ||||||||||||||||||
| -b <path> | Just like -B this option enables the output of *.ctab files for Ballgown, but these files will be created in the provided directory <path> instead of the directory specified by the -o option. Note: adding the -e option is recommended with the -B/-b options, unless novel transcripts are still wanted in the StringTie GTF output. | ||||||||||||||||||
| -M <0.0-1.0> | Sets the maximum fraction of muliple-location-mapped reads that are allowed to be present at a given locus. Default: 0.95. | ||||||||||||||||||
| -x <seqid_list> | Ignore all read alignments (and thus do not attempt to perform transcript assembly) on the specified reference sequences. Parameter <seqid_list> can be a single reference sequence name (e.g. -x chrM) or a comma-delimited list of sequence names (e.g. -x 'chrM,chrX,chrY'). This can speed up StringTie especially in the case of excluding the mitochondrial genome, whose genes may have very high coverage in some cases, even though they may be of no interest for a particular RNA-Seq analysis. The reference sequence names are case sensitive, they must match identically the names of chromosomes/contigs of the target genome against which the RNA-Seq reads were aligned in the first place. | ||||||||||||||||||
| -u | Turn off multi-mapping correction. In the default case this correction is enabled, and each read that is mapped in n places only contributes 1/n to the transcript coverage instead of 1. | ||||||||||||||||||
| --ref/--cram-ref | for CRAM input files, the reference genome sequence can be provided as a multi-FASTA file the same chromosome sequences that were used when aligning the reads. This option is optional but recommended as StringTie can make use of some alignment/junction quality data (mismatches around the junctions) that can be more accurately assessed in the case of CRAM files when the reference genome sequence is also provided. | ||||||||||||||||||
| --merge |
Transcript merge mode. This is a special usage mode of StringTie, distinct from the assembly usage mode described above. In the merge mode, StringTie takes as input a list of GTF/GFF files and
merges/assembles these transcripts into a non-redundant set of transcripts. This mode is used in the new differential analysis
pipeline to generate a global, unified set of transcripts (isoforms) across multiple RNA-Seq samples.
If the -G option (reference annotation) is provided, StringTie will assemble the transfrags from the input GTF files with the reference transcripts. The following additional options can be used in this mode:
|
Back to top
Input files
StringTie takes as input a SAM, BAM or CRAM file sorted by coordinate (genomic location). This file should contain spliced RNA-seq read alignments such as the ones produced by TopHat or HISAT2, or STAR. The TopHat output is already sorted, but the SAM ouput from other aligners should be sorted using the samtools program:
samtools sort -o alnst.sorted.bam alns.sam
The file resulted from the above command (alns.sorted.bam) can be used as input file for StringTie.
Any SAM record with a spliced alignment (i.e. having a read alignment across at least one junction) should have the XS tag (or the ts tag, see below) which indicates the transcription strand, the genomic strand from which the RNA that produced the read originated. TopHat and HISAT2 alignments already include this tag, but for other read mappers one should check that this tag is also included for spliced alignment records. For example the STAR aligner should be run with the option --outSAMstrandField intronMotif in order to generate this tag.
The XS tags are not necessary in the case of long RNA-seq reads aligned with minimap2 using the -ax splice option. minimap2 adds the ts tags to spliced alignments to indicate the transcription strand (albeit in a different manner than the XS tag) and StringTie can make use of the ts tag as well if the XS tag is missing.
When CRAM files are used as input, the reference genomic sequences can be provided with the --ref (--cram-ref) option
as a multi-FASTA file with the same chromosome sequences to which the RNA-seq reads were aligned. This is optional but recommended because StringTie
can better estimate the quality of some spliced alignments (e.g. keeping track of mismatches around junctions) and such data can be retrieved
in the case of some CRAM files only when the reference genome sequence is also provided.
Long reads assembly mode (-L)
The -L option must be used when the input alignment file contains (sorted) spliced alignments of long read RNA-seq or cDNA reads. Such alignments can be produced by `minimap2` with the `-ax splice` option, which also generates the necessary `ts` tag to indicate the transcription strand. As mentioned above such `minimap2` alignment files must be first position-sorted by before they can be processed by StringTie.
Mixed reads assembly mode (--mix)
With the --mix option StringTie can process both short RNA-Seq read alignments (usually aligned with TopHat2, HISAT2 or STAR) and long RNA-Seq/cDNA read alignments (usually aligned with minimap2). With this option two input files are expected, with the two types of alignments provided as two separate input files (BAM/CRAM format) in a specific order in the command line:
- the first input file should have the short read alignments
- the second input file is the one with the long read alignments
Both alignment files must be sorted by genomic location. The generic command line in this case becomes:
stringtie [-o <output.gtf>] --mix [other_options] <short_read_alns.bam> <long_read_alns.bam>
The regular options include a reference annotation (-G) and an output file name (-o), so a more realistic
command line example would look like this:
stringtie --mix -G $ref/hg38_annotation.gff -o $sample1dir/out/transcripts.gtf \ $sample1dir/short_reads.cram $sample1dir/long_reads.bam
Reference annotation transcripts (-G)
A reference annotation file in GTF or GFF3 format can be provided to StringTie using the -G option which can be used as 'guides' for the assembly process and help improve the transcript structure recovery for those transcripts.
NOTE: we highly recommend that you provide annotation if you are analyzing a genome that is well annotated, such as human, mouse, or other model organisms.
Note that when a reference transcript is fully covered by input read alignments, the original transcript ID from the reference annotation file will be shown in StringTie's output file in the reference_id GTF attribute for that assembled transcript. Output transcripts lacking the reference_id attribute can be considered "novel" transcript structures with respect to the provided reference annotation.
Expression estimation mode (-e)
When the -e option is used, the reference annotation file -G is a required input and StringTie will not attempt to assemble the input read alignments but instead it will only estimate the expression levels of the "reference" transcripts provided in the -G file.
With this option, no "novel" transcript assemblies (isoforms) will be produced, and read alignments not overlapping any of the given reference transcripts will be ignored, which may provide a considerable speed boost when the given set of reference transcripts is limited to a set of target genes for example.
Back to top
Output files
Main Outputs- Stringtie's main output is a GTF file containing the assembled transcripts
- Gene abundances in tab-delimited format
- Fully covered transcripts that match the reference annotation, in GTF format
- Files (tables) required as input to Ballgown, which uses them to estimate differential expression
- In merge mode, a merged GTF file from a set of GTF files
1. StringTie's primary GTF output The primary output of StringTie is a Gene Transfer Format (GTF) file that contains details of the transcripts that StringTie assembles from RNA-Seq data. GTF is an extension of GFF (Gene Finding Format, also called General Feature Format), and is very similar to GFF2 and GFF3. The field definitions for the 9 columns of GTF output can be found at the Ensembl site here. The following is an example of a transcript assembled by StringTie as shown in a GTF file (scroll right within the box to see the full field contents):
seqname source feature start end score strand frame attributes
chrX StringTie transcript 281394 303355 1000 + . gene_id "ERR188044.1"; transcript_id "ERR188044.1.1"; reference_id "NM_018390"; ref_gene_id "NM_018390"; ref_gene_name "PLCXD1"; cov "101.256691"; FPKM "530.078918"; TPM "705.667908";
chrX StringTie exon 281394 281684 1000 + . gene_id "ERR188044.1"; transcript_id "ERR188044.1.1"; exon_number "1"; reference_id "NM_018390"; ref_gene_id "NM_018390"; ref_gene_name "PLCXD1"; cov "116.270836";
...
Description of each column's values:
- seqname: Denotes the chromosome, contig, or scaffold for this transcript. Here the assembled transcript is on chromosome X.
- source: The source of the GTF file. Since this example was produced by StringTie, this column simply shows 'StringTie'.
- feature: Feature type; e.g., exon, transcript, mRNA, 5'UTR).
- start: Start position of the feature (exon, transcript, etc), using a 1-based index.
- end: End position of the feature, using a 1-based index.
- score: A confidence score for the assembled transcript. Currently this field is not used, and StringTie reports a constant value of 1000 if the transcript has a connection to a read alignment bundle.
- strand: If the transcript resides on the forward strand, '+'. If the transcript resides on the reverse strand, '-'.
- frame: Frame or phase of CDS features. StringTie does not use this field and simply records a ".".
- attributes: A semicolon-separated list of tag-value pairs, providing additional information about each
feature. Depending on whether an instance is a transcript or an exon and on whether the
transcript matches the reference annotation file provided by the user, the content of the attributes field will
differ. The following list describes the possible attributes shown in this column:
- gene_id: A unique identifier for a single gene and its child transcript and exons based on the alignments' file name.
- transcript_id: A unique identifier for a single transcript and its child exons based on the alignments' file name.
- exon_number: A unique identifier for a single exon, starting from 1, within a given transcript.
- reference_id: The transcript_id in the reference annotation (optional) that the instance matched.
- ref_gene_id: The gene_id in the reference annotation (optional) that the instance matched.
- ref_gene_name: The gene_name in the reference annotation (optional) that the instance matched.
- cov: The average per-base coverage for the transcript or exon.
- FPKM: Fragments per kilobase of transcript per million read pairs. This is the number of pairs of reads aligning to this feature, normalized by the total number of fragments sequenced (in millions) and the length of the transcript (in kilobases).
- TPM: Transcripts per million. This is the number of transcripts from this particular gene normalized first by gene length, and then by sequencing depth (in millions) in the sample. A detailed explanation and a comparison of TPM and FPKM can be found here, and TPM was defined by B. Li and C. Dewey here.
-A <gene_abund.tab> option, it returns
a file containing gene abundances. The tab-delimited gene abundances output file
has nine fields; here is an example of a gene abundance file produced
by StringTie using reference annotation:
Gene ID Gene Name Reference Strand Start End Coverage FPKM TPM
NM_000451 SHOX chrX + 624344 646823 0.000000 0.000000 0.000000
NM_006883 SHOX chrX + 624344 659411 0.000000 0.000000 0.000000
...
- Column 1 / Gene ID: The gene identifier comes from the reference annotation
provided with the
-Goption. If no reference is provided this field is replaced with the name prefix for output transcripts (-l). - Column 2 / Gene Name: This field contains the gene name in the reference
annotation provided with the
-Goption. If no reference is provided this field is populated with '-'. - Column 3 / Reference: Name of the reference sequence that was used in the alignment of the reads. Equivalent to the 3rd column in the .SAM alignment.
- Column 4 / Strand: '+' denotes that the gene is on the forward strand, '-' for the reverse strand.
- Column 5 / Start: Start position of the gene (1-based index).
- Column 6 / End: End position of the gene (1-based index).
- Column 7 / Coverage: Per-base coverage of the gene.
- Column 8 / FPKM: normalized expression level in FPKM units (see previous section).
- Column 9 / TPM: normalized expression level in RPM units (see previous section).
3. Fully covered transcripts matching the reference annotation transcripts (in GTF format)
If StringTie is run with the -C <cov_refs.gtf> option
(requires -G <reference_annotation>), it returns a file
with all the transcripts in the reference annotation that are fully
covered, end to end, by reads. The output format is a GTF file as described above.
Each line of the GTF is corresponds to a gene or transcript in the reference annotation.
4. Ballgown Input Table Files
If StringTie is run with the -B option, it
returns a Ballgown input table file, which contains coverage
data for all transcripts. The output table files are
placed in the same directory as the main GTF output. These tables
have these specific names: (1) e2t.ctab, (2) e_data.ctab, (3) i2t.ctab, (4)
i_data.ctab, and (5) t_data.ctab. A detailed description of each of
these five required inputs to Ballgown can be found on the Ballgown
site, at this link.
5. Merge mode: Merged GTF
If StringTie is run with the --merge option, it
takes as input a list of GTF/GFF files and merges/assembles
these transcripts into a non-redundant set of transcripts. This
step creates a uniform set of transcripts for all samples to
facilitate the downstream calculation of differentially
expressed levels for all transcripts among the different
experimental conditions. Output is a merged GTF file with all
merged gene models, but without
any numeric results on coverage, FPKM, and
TPM. Then, with this merged GTF, StringTie can re-estimate abundances by
running it again with the -e option on the original
set of alignment files, as illustrated in the figure below.
Evaluating transcript assemblies
A simple way of getting more information about the transcripts assembled by StringTie (summary of gene and transcript counts, novel vs. known etc.), or even performing basic tracking of assembled isoforms across multiple RNA-Seq experiments, is to use the gffcompare program. Basic usage information and download options for this program can be found on the GFF utilities page.
Back to topDifferential expression analysis
Together with HISAT and Ballgown, StringTie can be used for estimating differential expression across multiple RNA-Seq samples and generating plots and differential expression tables as described in our protocol paper.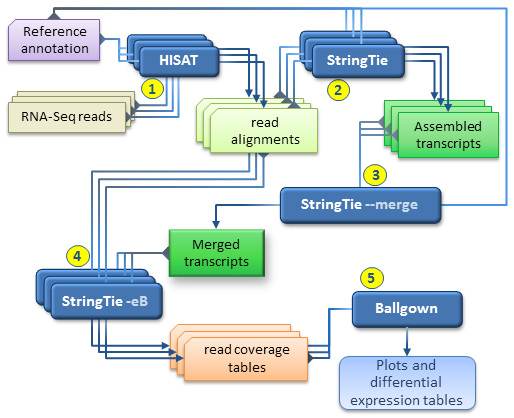
Our recommended workflow includes the following steps:
- for each RNA-Seq sample, map the reads to the genome with HISAT2 using the --dta option. It is highly recommended to use the reference annotation information when mapping the reads, which can be either embedded in the genome index (built with the --ss and --exon options, see HISAT2 manual), or provided separately at run time (using the --known-splicesite-infile option of HISAT2). The SAM output of each HISAT2 run must be sorted and converted to BAM using samtools as explained above.
- for each RNA-Seq sample, run StringTie to assemble the read alignments obtained in the previous step; it is recommended to run StringTie with the -G option if the reference annotation is available.
- run StringTie with --merge in order to generate a non-redundant set of transcripts observed in any of the RNA-Seq samples assembled previously. The stringtie --merge mode takes as input a list of all the assembled transcripts files (in GTF format) previously obtained for each sample, as well as a reference annotation file (-G option) if available.
- for each RNA-Seq sample, run StringTie using the -B/-b and -e options in order to
estimate transcript abundances and generate read coverage tables for Ballgown. The
-e option is not required but recommended for this run in order to produce more accurate abundance estimations of the input transcripts. Each StringTie run in this step will take as input the sorted read alignments (BAM file) obtained in step 1 for the corresponding sample and the -G option with the merged transcripts (GTF file) generated by stringtie --merge in step 3. Please note that this is the only case where the -G option is not used with a reference annotation, but with the global, merged set of transcripts as observed across all samples. (This step is the equivalent of the Tablemaker step described in the original Ballgown pipeline.) - Ballgown can now be used to load the coverage tables generated in the previous step and perform various statistical analyses for differential expression, generate plots etc.
An alternate, faster differential expression analysis workflow can be pursued if there is no interest in novel isoforms (i.e. assembled
transcripts present in the samples but missing from the reference annotation), or if only a well known set of transcripts of
interest are targeted by the analysis.
This simplified protocol has only 3 steps (depicted below) as it bypasses the individual assembly of each RNA-Seq sample and the
"transcript merge" step.
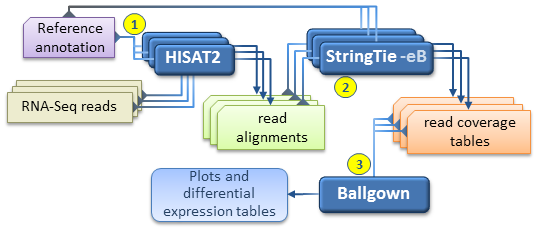 This simplified workflow attempts to directly estimate and analyze the expression of a known set of transcripts as given in the reference annotation file.
This simplified workflow attempts to directly estimate and analyze the expression of a known set of transcripts as given in the reference annotation file.
The R package IsoformSwitchAnalyzeR can be used to
assign gene names to transcripts assembled by StringTie, which can be particularly helpful in cases where StringTie could not
perform this assignment unambigiously.
The importIsoformExpression() + importRdata() function of the package can be
used to import the expression and annotation data into R.
During this import the package will attempt to clean up and recover isoform annotations where possible. From the resulting
switchAnalyzeRlist object, IsoformSwitchAnalyzeR can detect isoform switches along with predicted
functional consequences.
The extractGeneExpression() function can be used to get a gene expression (read count) matrix for analysis with other tools.
More information and code examples can be found
here.
Using StringTie with DESeq2 and edgeR
DESeq2 and edgeR are two popular Bioconductor packages for analyzing differential expression, which take as input a matrix of read counts mapped to particular genomic features (e.g., genes). We provide a Python script (prepDE.py, or the Python 3 version: prepDE.py3 ) that can be used to extract this read count information directly from the files generated by StringTie (run with the -e parameter).
prepDE.py derives hypothetical read counts for each transcript from the coverage values estimated by StringTie for each transcript, by using this simple formula:
reads_per_transcript = coverage * transcript_len / read_len
There are two ways to provide input to the prepDE.py script:
- one option is to provide a path to a directory containing all sample sub-directories, with the same structure as
the ballgown directory in the StringTie protocol paper in preparation for Ballgown.
By default (no -i option), the script is going to look in the current directory for all sub-directories having .gtf files in them, as in this example:
./sample1/sample1.gtf ./sample2/sample2.gtf ./sample3/sample3.gtf - Alternatively, one can provide a text file listing sample IDs and their respective paths (sample_lst.txt).
Usage: prepDE.py [options]
generates two CSV files containing the count matrices for genes and transcripts, using the coverage values found in the output of stringtie -e
| -h, --help | show this help message and exit |
| -i INPUT, --input=INPUT, --in=INPUT | a folder containing all sample sub-directories, or a text file with sample ID and path to its GTF file on each line [default: . ] |
| -g G | where to output the gene count matrix [default: gene_count_matrix.csv] |
| -t T | where to output the transcript count matrix [default: transcript_count_matrix.csv] |
| -l LENGTH, --length=LENGTH | the average read length [default: 75] |
| -p PATTERN, --pattern=PATTERN | a regular expression that selects the sample subdirectories |
| -c, --cluster | whether to cluster genes that overlap with different gene IDs, ignoring ones with geneID pattern (see below) |
| -s STRING, --string=STRING | if a different prefix is used for geneIDs assigned by StringTie [default: MSTRG] |
| -k KEY, --key=KEY | if clustering, what prefix to use for geneIDs assigned by this script [default: prepG] |
| --legend=LEGEND | if clustering, where to output the legend file mapping transcripts to assigned geneIDs [default: legend.csv] |
These count matrices (CSV files) can then be imported into R for use by DESeq2 and edgeR (using the DESeqDataSetFromMatrix and DGEList functions, respectively).
Protocol: Using StringTie with DESeq2
Given a list of GTFs, which were re-estimated upon merging, users can follow the below protocol to use DESeq2 for differential expression analysis. To generate GTFs from raw reads follow the StringTie protocol paper (up to the Ballgown step).
As described above, prepDE.py either accepts a .txt (sample_lst.txt) file listing the sample IDs and the GTFs' paths or by default expects a ballgown directory produced by StringTie run with the -B option. The following steps leads us through generating count matrices for genes and transcripts, importing this data into DESeq2, and conducting some basic analysis.
- Generate count matrices using prepDE.py
- Assuming default ballgown directory structure produced by StringTie
$ python prepDE.py - List of GTFs sample IDs and paths (sample_lst.txt)
$ python prepDE.py -i sample_lst.txt
- Assuming default ballgown directory structure produced by StringTie
- Install R and DESeq2. Upon installing R, install DESeq2 on R:
> source("https://bioconductor.org/biocLite.R")
> biocLite("DESeq2") - Import DESeq2 library in R
> library("DESeq2") - Load gene(/transcript) count matrix and labels
> countData <- as.matrix(read.csv("gene_count_matrix.csv", row.names="gene_id"))
> colData <- read.csv(PHENO_DATA, sep="\t", row.names=1)
Note: The PHENO_DATA file contains information on each sample, e.g., sex or population. The exact way to import this depends on the format of the file. - Check all sample IDs in colData are also in CountData and match their orders
> all(rownames(colData) %in% colnames(countData))[1] TRUE> countData <- countData[, rownames(colData)][1] TRUE
> all(rownames(colData) == colnames(countData)) - Create a DESeqDataSet from count matrix and labels
> dds <- DESeqDataSetFromMatrix(countData = countData,
colData = colData, design = ~ CHOOSE_FEATURE) - Run the default analysis for DESeq2 and generate results table
> dds <- DESeq(dds)
> res <- results(dds) - Sort by adjusted p-value and display
> (resOrdered <- res[order(res$padj), ])
Back to top
Assembling super-reads
Although StringTie is primarily a genome-guided approach, it can borrow algorithmic techniques from de novo genome assembly to help with transcript assembly. Its input can include not only the spliced read alignments used by reference-based assemblers, but also longer contigs that it assembles de novo from unambiguous, non-branching parts of a transcript. If your RNA-Seq data is paired, you could use our method to reconstruct the RNA-Seq fragments from their end sequences, which we call super-reads. This is an optional step for preparing the alignments for the input of StringTie, but it can improve the accuracy of the transcriptome assembly. To use this step you will first need to install the MaSuRCA genome assembler. The super-read module from MaSuRCA extends every read in both directions as long as this extension is unique. The superreads.pl script that we provide here identifies pairs of reads that belong to the same super-read, and extracts the sequence containing the pair plus the sequence between them; i.e., the entire sequence of the original DNA fragment. Thus for example if the original RNA-seq data comprised paired 100-bp reads from a 300-bp fragment library, these steps will convert many of those pairs into single, 300-bp super-reads.The usage of the superreads.pl script is documented below.
Usage: superreads.pl <pair_read1_fastq> <pair_read2_fastq> <masurca_directory> [options]*
Arguments:
The first two arguments of the superreads.pl script are files in the fastq format containing the the sequences of the first and second read in each fragment, respecitvely. They can either plain text fastq files or compressed (with gzip or bzip2) files The third argument represents the directory where the MaSuRCA package was installed on your system.
Options:
| -t <num_threads> | Sets the number of threads to use. Default: 10. |
| -j <jf_size> | MaSuRCA requires the Jellyfish program to run, and this parameter sets the Jellyfish hash size. Please see the MaSuRCA documentation for more information about how to choose this parameter. Default: 2500000000. |
| -s <step> | As it progresses, the superreads.pl script prints the steps it successfully completed. If for any reason, the assembly process is stopped, you don't need to redo all the successfully completed steps, and you can restart the script at the first step it didn't complete. Default: 1. |
| -r <paired_read_prefix> | Sets the prefix for the paired reads as required by MaSuRCA. Default: pe. |
| -f <fragment_size> | Specifies the mean library insert length. Default: 300. |
| -d <standard_deviation> | Specifies the standard deviation of the library insert length. If the standard deviation is not known, set it to approximately 15% of the mean. Default: 45. |
| -l <super_reads_file_name> | Specifies the name for the assembled super-reads file. Default: LongReads.fq. |
| -u <not_assembled_reads_prefix> | Specifies the prefix for the unassembled reads file names. By default it appends ".notAssembled.fq.gz" to the initial paired files. |
The output files of the superreads.pl script (the assembled super-reads file, and the two files containing the unassembled paired reads) can be then aligned to a reference genome with your read mapper of preference. For instance, you can align them with TopHat2 like this:
tophat [options]* <genome_index_base> \ PE_reads_1.notAssembled.fq.gz,LongReads.fq PE_reads_2.notAssembled.fq.gz
Back to top

