Q & A ...#
Q: How much does LiftOn improve over Liftoff and miniprot?
Here is one example of improvement over human annotation lift-over from GRCh38 to T2T-CHM13.
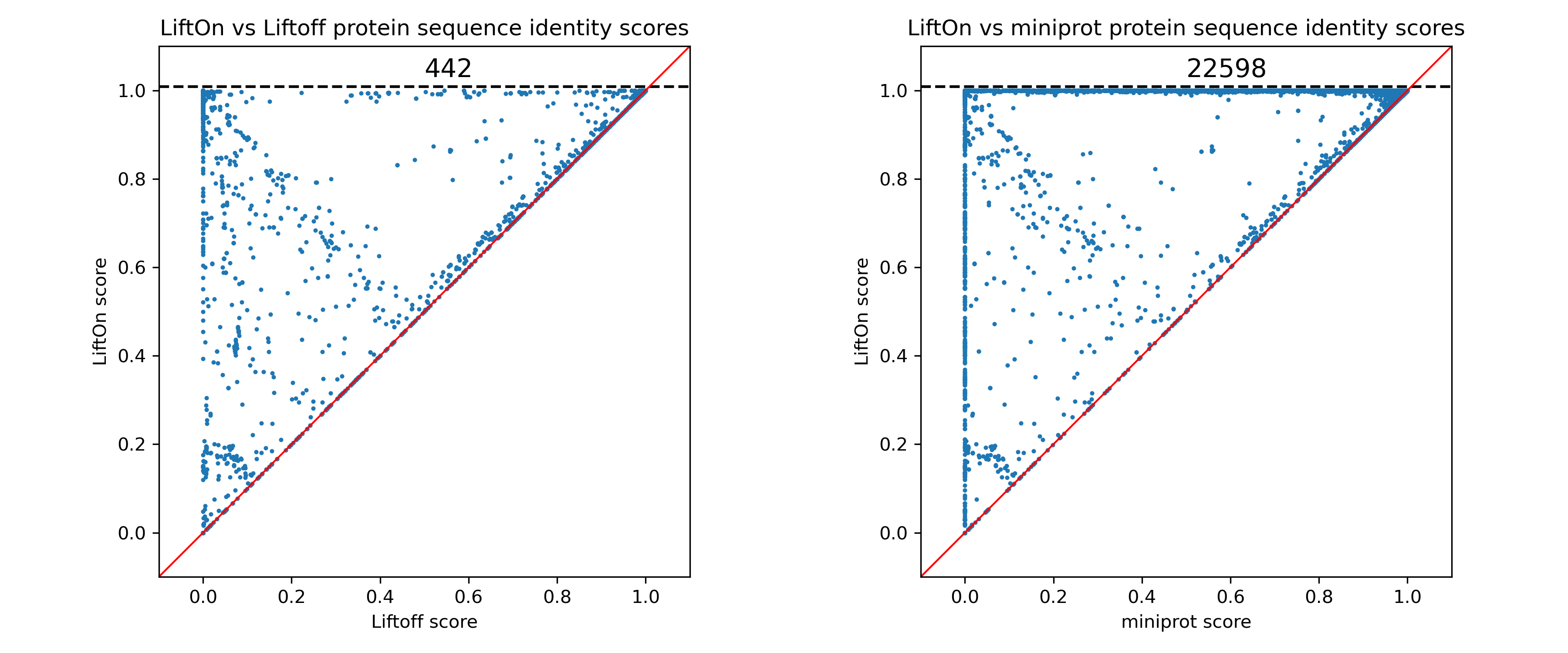
Each dot represents a protein-coding transcript. If it is above the x=y line, it indicates that the LiftOn annotation possesses a higher protein sequence identity score and corresponds to a longer protein that aligns with the proteins in the reference annotation.
In the LiftOn versus Liftoff comparison (Figure above, left), 2,075 transcripts exhibit higher protein sequence identity, with 460 achieving 100% identity. Similarly, the LiftOn versus miniprot comparison (Figure above, right) discloses better matches for 30,276 protein-coding transcripts, improving 22,616 to identical status relative to the reference.
In summary, LiftOn effectively corrects quite a few protein-coding transcripts during human lift-over. The improvement is even more significant when it comes to more distant species!
Check out the Same species lift-over section, Closely related species lift-over section, and Distantly related species lift-over section for more details.