

Bos taurus assembly
The genome of the domestic cow, Bos taurus, was sequenced using a mixture of hierarchical and whole-genome shotgun sequencing methods. The sequences were assembled by the Center for Bioinformatics and Computational Biology at University of Maryland (CBCB) using the Celera Assembler [1]. Two major releases have been made available to the public: UMD2 (April 2009) [2] and UMD3.0 (August 2009) [3]. A minor release UMD3.1 was made available to the public in December 2009. The UMD3.1 assembly is identical in almost all respects to UMD3.0. The only changes are in the AGP file deposited at GenBank. All contigs and chromosomes are unchanged.
The first release, UMD2, published in Genome Biology in April 2009 [4], assembled 35.62 million reads into a 2.85 billion bp genome out of which 2.61 billion (91%) bp were placed on chromosomes. Celera Assembler version 4.8 was used to assemble the Baylor College of Medicine (BCM) reads [7],.
The second release, UMD3 (described here) assembled 36.82 million reads into a 2.649 billion bp genome out of which 2.640 billion (99%) bp were placed on chromosomes. Celera Assembler version 5.2 was used to assemble all the genomic reads available in the NCBI Trace Archive (TA) [5]: 35.62 million BCM reads plus 1.2 million reads generated by other sequencing centers.
Post-processing algorithms that utilize paired-end sequence information, marker mapping and synteny with the human genome were used in order to detect and correct errors. Using extensive marker and synteny data, we were able to anchor almost all of the assembled genome onto chromosomes.
UMD3 marks a significant improvement over UMD2 with many fewer gaps, smaller gaps, and approximately 3% more sequence placed on chromosomes. Both assembly are accessible online for FTP download [2],[3] or BLAST search [12],[13].
UMD3 is available in GenBank under the following accession numbers:
- Project: DAAA00000000
- Chromosomes: GK000001:GK000030
- Contigs: DAAA02000001:DAAA02075770
- Placed scaffolds: GJ060423:GJ063645
- Unplaced scaffolds: GJ057137:GJ060422
Annotation for UMD3 generated by the NCBI Gnomon pipeline is available (since Feb 2010) at the CBCB ftp site or at the the CCB ftp site, which also contains the assembly. See the README file at this site for details. Note that UMD3.1 is identical to UMD3.0; the only changes are internal files at GenBank, while all contigs and scaffolds remain the same. Also note that after the Salzberg lab moved to Johns Hopkins University, the ftp sites have all been moved from CBCB to the Center for Computational Biology (CCB) at JHU. The CCB site at JHU is more up-to-date.
The UMD 3.1 assembly, along with the new annotation, can also be browsed at the NAGRP GBrowse site, which also shows QTL markers.
In 2014, we identified a small number (175) of contigs that derived from bacterial and viral contaminants. All of these were originally unplaced and contained in the ChrU.fa file. We removed these from ChrU and created a minor new release, numbered 3.1.1.
This site focuses on the UMD3 assembly method and results.
1. Data
37.82 million Bos taurus reads were downloaded from NCBI Trace Archive [5].
About 1 million EST & PCR reads were filtered out, leaving 36.82 million whole genome shotgun (WGS), BAC-based shotgun, BAC-end sequencing & BAC-finishing reads to be assembled.
Out of the total set, 35.62 million reads were sequenced by Baylor College of Medicine (BCM) [7] while another 1.20 million were sequenced by other centers [8]
The table below contains a count of the reads by center and read type.
| CENTER CODE | READ TYPE | READ COUNT |
|---|---|---|
| Baylor College of Medicine | Whole Genome Shotgun | 24,863,599 |
| Baylor College of Medicine | BAC Based Shotgun | 10,748,529 |
| NIH Intramural Sequencing Center | BAC Based Shotgun | 737,900 |
| British Columbia Cancer Agency Genome Sciences Centre | BAC End Sequencing | 12,5597 |
| University of Illinois at Urbana-Champaign | BAC End Sequencing | 114,753 |
| The Institute for Genome Research | BAC End Sequencing | 65,171 |
| Genoscope | BAC End Sequencing | 53,556 |
| Embrapa Genetic Resources and Biotechnology | Whole Genome Shotgun | 26,246 |
| USDA, ARS, Beltsville Agricultural Research Center | BAC End Sequencing | 25,454 |
| Baylor College of Medicine | BAC End Sequencing | 16,892 |
| Embrapa Genetic Resources and Biotechnology | BAC End Sequencing | 16,787 |
| University of Oklahoma Norman Campus, Advanced Center for Genome Technology | BAC Based Shotgun | 15,150 |
| The Institute for Genomic Research, Traces generated at JCVIJTC | BAC End Sequencing | 10,651 |
| University of Oklahoma Norman Campus, Advanced Center for Genome Technology | BAC Finishing | 151 |
| Washington University, Genome Sequencing Center | BAC End Sequencing | 49 |
| TOTAL | 36,820,485 |
The reads were analyzed and pre-processed prior to the assembly.
Several problems were noticed with the reads:
1. 0.65 million reads were missing quality values (henceforth referred to as "quality-less" reads).
2. The reads were tagged as coming from 25,312 libraries; some libraries were very fragmented and contained only a few reads.
3. Some libraries had incorrect library insert size estimates (e.g.: WGS libraries labeled as BAC ends)
4. Some traces were missing or had incomplete vector trimming coordinates.
The reads were re-trimmed using the Lucy software [16]. Lucy is a program for quality trimming and vector removal. It takes as input the vector and splice site used in the sequencing process. For each library we identified the linker and splice site sequences based on high frequency k-mers. The cloning vectors were identified based on alignments to the UniVec database. Lucy was run on all reads and the vector trimming coordinates were extracted from the Lucy output.
2. Preliminary assembly
The preliminary assembly was based on the 36.17 million quality reads. The overlap-based trimming method (OBT) and the Lucy vector trimming coordinates were used. All the BAC-based shotgun reads were labeled as non-random so that the assembler would not treat these regions as repeats.
Celera assembled 35.34 million reads into 66,141 scaffolds, 120,461 contigs and 269,031 degenerates.
The longest scaffold was 26.04 million bp and the average length was 40.86 kilobases.
The longest contig was 269,030 bp and the average length was 22,430 bp.
3.72 million reads were left as singletons (unused) and 421,370 reads were deleted (too short or chimeric).
We then used this preliminary assembly for re-trimming the reads, library processing and contamination search.
1. The good part of each read is the "clear range." We used the OBT clear ranges for the quality reads.
The 0.65 million quality-less reads were aligned to the assembled contigs using the MUMmer software [10];
the aligned intervals were set as the read clear ranges.
2. The 25,312 libraries were consolidated into 344 libraries by grouping reads from the same sequencing center, sequencing type and similar insert size estimates.
3. For contamination search, we aligned all the contigs and degenerates to a database of E. coli, UniVec vectors [11] and 100 other common RefSeq vector sequences.
40,699 reads were identified as contaminants and deleted from the input set.
3. Final assembly
The final assembly was based on both the quality and quality-less reads. Since reads had already been trimmed, the overlap-based trimming method (OBT) was disabled. The contaminant reads were deleted and the BAC-based shotgun reads were again labeled as non-random.
Celera Assembler assembled 35.97 million reads into 39,978 scaffolds, 90,135 contigs and 251,413 degenerates
The longest scaffold was 33.9 million bp while the average was 66,940 bp.
The longest contig was 1.16 million bp while the average was 29,690 bp.
3.63 million reads were left as singletons.
The final assembly marks a significant improvement over the preliminary one.
4. Assembly processing
4.1. Contaminant search
We used the same contaminant databases and alignment parameters as for the preliminary assembly processing.
Some contamination was still present: 156 contigs, 152 scaffolds and 4,105 reads were identified as contaminants and deleted.
12 long contigs contained partial contaminant sequences which we trimmed off.
4.2. Mapping the assembly onto the chromosomes
4.2.1. Marker based mapping
A BAC-end fingerprint marker map was used for placing the assembly onto the chromosomes.
The BAC-end markers were aligned to the assembly using the MUMmer software.
A marker was considered as matching a chromosome if 90% of the marker sequence aligned with at least 95% identity.
Most markers had multiple alignments and since the mapping had to the unique we only used the best score alignment (which could actually be incorrect).
Overall 107,899 out of the 126,013 markers aligned at 2,640 scaffolds, 31,407 contigs and 562 degenerates.
37,338 scaffolds had no markers, 1,595 scaffolds had one marker while 1,045 had more than two markers aligned to them.
We used the marker alignments to detect misassembled sequences.
The hypothesis was that there is a very low chance to have multiple markers from multiple chromosomes aligned to the same sequnce in near proximity of one another.
To confirm the hypothesis we computed the read and mate pair coverage for the possibly misassembled regions.
Based on a low read coverage and significantly large number of unhappy mates, 15 scaffolds were confirmed as misassembled and split.
The assignment of assembled sequences to the chromosomes was done based on a best alignment & majority rule.
The Interquartile Range method was used to filter out suspicious markers and compute the scaffold/degenerate positions on the chromosomes.
The Least Square method was used to compute the orientation of the scaffolds/degenerates that had multiple markers aligned to them.
Orientation was considered forward if the slope value was positive, reverse if the slope was negative and unknown if the slope was close to 0.
4.2.3. Synteny based mapping
We managed to place and oriented additional assembled sequences on the chromosomes using cow-human alignments. All the assembled sequences were aligned to the 24 human chromosomes using MUMmer. 9,914 scaffolds & 16,527 degenerates aligned to human, significantly more than the 2,640 scaffolds & 562 degenerates that had markers. The position/orientation on the chromosomes of the aligned sequences were inferred based on reliably placed neighbors (multiple marker aligned sequences).
4.2.4. Map refininment
Once the assembled sequences were assigned to chromosomes we used mate pair information to refine placements. We identified questionably placed or unplaced sequences linked to the reliably placed sequences and tried to fit them into the appropriate gaps. Genome scaffolding with iterative least squares method was used to recompute the positions and orienteations of contigs on chromosomes.
4.3. Haplotype variant removal
While evaluating the assembly for correctness, we found many examples of contigs placed along the chromosomes that aligned nearly identically with nearby contigs. When the two copies of each chromosome in a diploid genome diverge sufficiently, a genome assembler will be unable to merge the reads coming from the two haplotypes into a single consensus sequence. Instead, it partitions the reads into two separate contigs, one of which must be retained and the other one removed.
To detect and correct the haplotype variant problem, we aligned each contig to all contigs nearby.
Those that aligned with >97% identity for >90% of their length or had less than 2,000 unique bp were removed from the assembly and placed in a separate haplotype variants file.
This procedure removed 40,611 contigs and degenerates, totaling approximately 59.95 million bp of sequence.
5. Assembly Summary
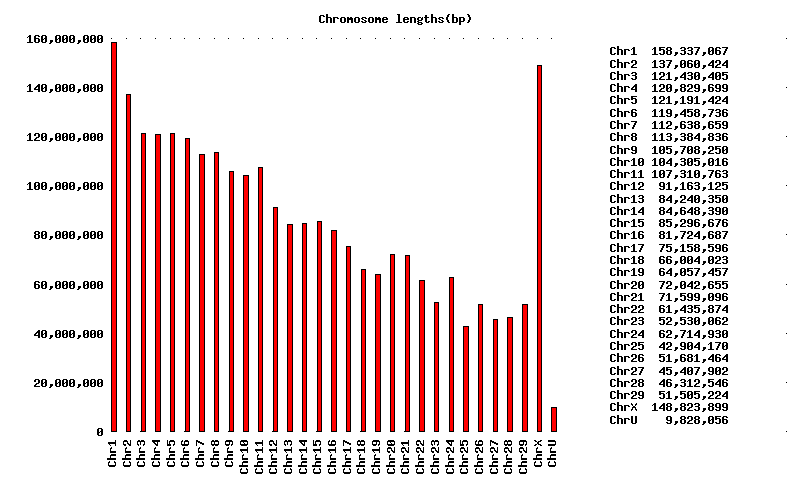
The Bos taurus UMD3 assembly contains 2,640,169,142 bp in contigs placed on chromosomes and 9,499,556 bp in unplaced contigs.
There are 72,484 placed contigs and 3,286 unplaced contigs.
The contig N50 length based on a genome size of 2.47 billion bp is 103,785 bp.
The largest contig contains 1,160,130 bp.
Below is a summary of the processed UMD3 assembly results. A detailed explanation of the results is available on the assembly FTP site [6].
| COUNT | <2Kbp | >=2Kbp | MIN | MAX | MEAN | MED | N50 | SUM | |
|---|---|---|---|---|---|---|---|---|---|
| PLACED CONTIGS/DEGENERATES (CHR 1..29,X) | 72,484 | 20,862 | 51,622 | 65 | 1,160,130 | 36,424 | 12,941 | 103,785 | 2,640,169,142 |
| PLACED SCAFFOLDS | 3,223 | 1013 | 2,210 | 446 | 36,161,404 | 825,500 | 5,300 | 6,445,999 | 2,660,587,105 |
| UNPLACED CONTIGS/DEGENERATES | 3,286 | 2,404 | 882 | 224 | 179,692 | 2,890 | 1,338 | 5,425 | 9,499,556 |
| PLACED & UNPLACED CONTIGS/DEGENERATES | 75,770 | 23,266 | 52,504 | 65 | 1,160,130 | 34,969 | 11,207 | 103,840 | 2,649,668,698 |
| HAPLOTYPE VARIANT CONTIGS/DEGENERATES | 40,605 | 36,984 | 3,621 | 263 | 51,828 | 1,472 | 1,205 | 1,369 | 59,771,100 |
| SHORT UNPLACED DEGENERATES | 224,933 | 224,933 | 0 | 65 | 1,996 | 972 | 983 | 990 | 218,837,572 |
| ChrY CONTIGS/DEGENERATES | 314 | 266 | 48 | 224 | 26,490 | 2,210 | 973 | 6,539 | 694,140 |
Compared with UMD2, UMD3 assembly contains 17.6 million bp more placed on chromosomes. Except for Chr2, Chr4, Chr14, Chr26 and Chr27, all the other chromosomes contain more sequence in the UMD3.0 version of the assembly. The largest size increase was noticed in ChrX which grew from 137.14 million bp UMD2 to 148.82 million bp in UMD3.
6. Human synteny map
The 30 Bos taurus chromosomes (UMD3 assembly) were aligned to the 24 Homo sapiens chromosomes (build 36.1) using the MUMmer software. (nucmer -l 12 -c 65 -g 1000 -b 1000)
Alignments which were less than 200bp or not reciprocal best hits were filtered out. (delta-filter -l 200 -1)
The alignments were grouped and sorted accoding to the reference and the position on the reference. For each alignment, from 5 to 100 adjacent alignments on each side were inspected.
Two alignments to a given reference sequence were considered to agree if they have the same query, same query orientation and the distance between the reference intercepts less tha 1Mbp.
The percentage of the adjacent alignments that agree with a given alignment was computed.
If at least 25% of the adjacent alignments agreed, the window size would be increased by one and the the percent recomputed.
Otherwise the alignment would be considered unreliable and removed from the set.
The filtered alignments were clustered and maximal sets of non-conflicting adjacent alignments were identified.
| COUNT | <2Kbp | >=2Kbp | MIN | MAX | MEAN | MED | N50 | SUM | |
|---|---|---|---|---|---|---|---|---|---|
| all | 532,866 | 491,050 | 41,816 | 11 | 34,597 | 925 | 717 | 1,246 | 493,152,056 |
| reciprocal_best_hit_filtering | 392,789 | 352,805 | 39,984 | 11 | 34,597 | 1,015 | 749 | 1,376 | 398,713,561 |
| neighbor_based_filtering | 368,820 | 329,672 | 39,148 | 11 | 34,597 | 1,038 | 767 | 1,399 | 383,058,325 |
| neighbor_based_clustering | 323 | 0 | 323 | 191,406 | 86,757,102 | 8,304,707 | 3,413,785 | 19,750,702 | 2,682,420,539 |
We identifies 323 syntenic regions that correspond to 54 Homo sapiens - Bos taurus chromosome pairs.
7. References and links
- Celera Assembler software
- CCB UMD2 FTP site
- CCB UMD3 FTP site
- A whole-genome assembly of the domestic cow, Bos taurus Genome Biology, April 2009
- NCBI Trace Archive FTP
- UMD3 README file
- BCM Genome Project
- NCBI sequencing centers
- Lucy software
- MUMmer software
- NCBI UniVec database
- CBCB UMD2 & UMD3 MegaBLAST site
- CBCB UMD2 & UMD3 BLAST site
- NCBI CBCB Assembly Project
- Mappings of STS and RH markers onto UMD3 and Btau4.2


{kind=link}