predict#
The predict subcommand uses a trained OpenSpliceAI model to identify potential splice sites in user-provided DNA sequences. It accepts either a standalone FASTA file or both a FASTA and GFF (annotation) file to determine genomic coordinates for protein-coding genes. This command outputs BED files for donor and acceptor sites, optionally storing intermediate predictions in HDF5 or PyTorch format for later analysis.
Overview#
Once an OpenSpliceAI model is trained (via the train or transfer), the predict subcommand:
Loads a reference genome in FASTA and, optionally, a GFF file to focus on specific gene regions.
Splits large sequences into manageable chunks (
--split_fasta_threshold).One-hot encodes the sequence data with flanking regions (size determined by
--flanking-size).Runs inference on each chunk in a batch-wise manner (with a batch size derived from the flanking size).
Generates final BED files, separating donor and acceptor sites exceeding a user-defined score threshold.
Input Files#
FASTA File
A FASTA file containing sequences for which you want to predict splice sites. If a GFF file is also provided, only the genomic regions marked as “gene” in the GFF are extracted for prediction.
GFF File (Optional)
A GFF3 annotation file. If supplied, the tool extracts protein-coding gene regions from the FASTA for more targeted prediction.
Trained Model Checkpoint(s)
A PyTorch checkpoint (
.pt) file (state dictionary) from a trained OpenSpliceAI model, or a directory containing multiple of these files (for ensemble prediction, where it will return the average across all models).
Output Files#
Donor and Acceptor BED Files: Two BED files listing genomic coordinates (if GFF supplied, else absolute coordinates) and scores for splice sites. -
acceptor_predictions.bed-donor_predictions.bedIntermediate Files (depending on parameters): - HDF5 Files: One-hot-encoded sequences and/or raw predictions.
datafile.h5: Raw extracted sequencesdataset.h5: Batched input for predictionpredict.h5: Raw predictions from the model (if--predict-all)
Extracted FASTA: Refined versions of the input FASTA are created with overlapping regions to avoid missing predictions at chunk boundaries.
[name]_genes.fa: FASTA file containing only gene regions (if GFF provided)[name]_split.fa: Split FASTA file (if any sequence in the input FASTA exceeds the--split-threshold)
Usage#
usage: openspliceai predict [-h] [--model MODEL] --output-dir OUTPUT_DIR [--flanking-size FLANKING_SIZE] [--input-sequence INPUT_SEQUENCE]
[--annotation-file ANNOTATION_FILE] [--threshold THRESHOLD] [--predict-all] [--debug] [--hdf-threshold HDF_THRESHOLD]
[--flush-threshold FLUSH_THRESHOLD] [--split-threshold SPLIT_THRESHOLD] [--chunk-size CHUNK_SIZE]
optional arguments:
-h, --help show this help message and exit
--model MODEL, -m MODEL
Path to a PyTorch SpliceAI model file or "SpliceAI" for the default model
--output-dir OUTPUT_DIR, -o OUTPUT_DIR
Output directory to save the data
--flanking-size FLANKING_SIZE, -f FLANKING_SIZE
Sum of flanking sequence lengths on each side of input (i.e. 40+40)
--input-sequence INPUT_SEQUENCE, -i INPUT_SEQUENCE
Path to FASTA file of the input sequence
--annotation-file ANNOTATION_FILE, -a ANNOTATION_FILE
Path to GFF file of coordinates for genes
--threshold THRESHOLD, -t THRESHOLD
Threshold to determine acceptor and donor sites
--predict-all, -p Writes all collected predictions to an intermediate file (Warning: on full genomes, will consume much space.)
--debug, -D Run in debug mode (debug statements are printed to stderr)
--hdf-threshold HDF_THRESHOLD
Maximum size before reading sequence into an HDF file for storage
--flush-threshold FLUSH_THRESHOLD
Maximum number of predictions before flushing to file
--split-threshold SPLIT_THRESHOLD
Maximum length of FASTA entry before splitting
--chunk-size CHUNK_SIZE
Chunk size for loading HDF5 dataset
Examples#
Example: Predicting splice sites for human genes#
openspliceai predict \
--model /path/to/model_best.pt \
--input-sequence GRCh38.fa \
--annotation-file GRCh38.gff \
--flanking-size 400 \
--threshold 0.6 \
--output-dir ./prediction_output/
This command:
Extracts gene regions from
GRCh38.fausing coordinates inGRCh38.gff.Splits large sequences exceeding the default threshold into manageable chunks.
One-hot encodes each chunk with a 400-nt flanking region.
Loads the specified model, runs inference in turbo mode, and outputs predictions directly to BED files.
Writes donor and acceptor sites with probability > 0.6 into two BED files.
Example: Predicting all splice sites in sample#
openspliceai predict \
--model /path/to/model.pt \
--input-sequence sample.fa \
--flanking-size 10000 \
--predict-all \
--output-dir ./prediction_output/
This command:
Processes the entire
sample.fafile.Splits sequences longer than the default threshold into smaller chunks.
One-hot encodes each chunk with a 10,000-nt flanking region.
Loads the specified model and runs inference.
Writes all raw predictions to the output directory in HDF5 format.
Generates BED files for all predictions (technically, all predictions > 1e-6).
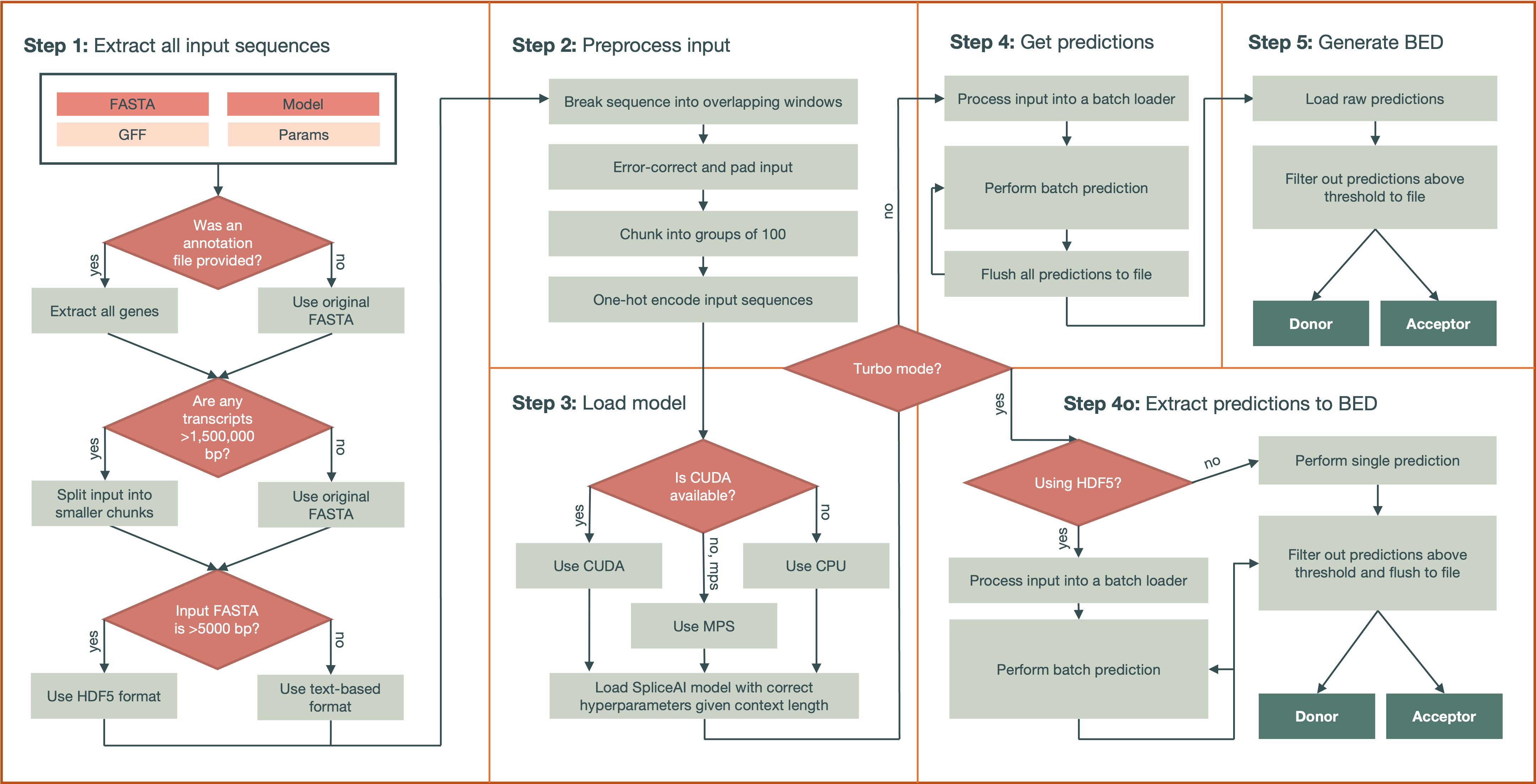
Processing Pipeline#
Sequence Extraction & Splitting
If a GFF file is provided, the subcommand creates a new FASTA containing only gene features (type = "gene").
Large sequences (exceeding
--split-threshold) are split into overlapping fragments to avoid missing predictions at boundaries.
One-Hot Encoding
Each sequence is padded with \(\frac{\text{flanking-size}}{2}\) on both ends using 'N's.
Overlapping windows of length 5,000 +
flanking_sizeare created, ensuring every base is covered.Sequences are grouped into chunks of size
--chunk-sizeto manage memory usage.
Model Loading
The specified PyTorch model checkpoint is loaded onto the best available device (GPU if available, otherwise CPU).
If multiple models or an ensemble directory is provided, predictions are averaged across all valid checkpoints (must be saved as a
.ptfile).
Batch Prediction
A DataLoader object feeds chunked, one-hot-encoded sequences to the model in batches, with the batch size set based on the flanking size.
Predictions are aggregated either: - Standard Mode (
--predict-all): Full predictions are stored in an HDF5 (or PyTorch) file before BED conversion. - Turbo Mode (default): Predictions are converted on-the-fly to BED entries without storing them fully.
BED File Generation
For each base in the sequence, the tool outputs donor or acceptor entries to two separate BED files if they exceed
--threshold.Coordinates are derived from: - GFF (if provided), yielding genomic coordinates. - FASTA headers, if no GFF is present (coordinates are then relative to the start of the FASTA entry).
The final result is two BED files: -
acceptor_predictions.bed-donor_predictions.bed
Workflow#
Conclusion#
The predict subcommand is the final step of the OpenSpliceAI pipeline, taking raw DNA sequences (FASTA) and a trained model and giving interpretable splice site predictions quickly and at big scales.