variant#
The variant subcommand evaluates the impact of genomic variants (SNPs and small INDELs) on splice sites by comparing model predictions on the reference (wild-type) sequence to predictions on the altered (mutant) sequence. It annotates a VCF file with “delta” scores for four events—acceptor gain, acceptor loss, donor gain, and donor loss—along with the relative position of each event. These delta scores reflect how much the mutation modifies the model’s predicted splice site strength and location.
Overview#
Similar to SpliceAI’s variant annotation approach (Jaganathan et al., 2019), the variant subcommand:
Parses a user-provided VCF file.
Retrieves the reference genome context from a FASTA file.
Loads a trained OpenSpliceAI model (PyTorch or Keras).
Predicts splice site probabilities for both wild-type and mutated sequences.
Computes the maximum change in donor or acceptor probability (delta scores) within a fixed window (default ±50 nt) around each variant for each event.
Outputs an annotated VCF, including delta scores and delta positions for each allele.
Input Files#
VCF File
A standard VCF file containing variants (SNPs, small insertions, and deletions).
Reference Genome (FASTA)
A FASTA file used to extract wild-type sequences. The subcommand checks that the reference allele in the VCF matches the reference genome.
Annotation File
A custom gene annotation file that defines the genomic regions to consider. Variants outside of annotated genes or too close to the chromosome ends (by default, within the flanking region) are skipped.
Trained Model Checkpoint(s)
A directory or file containing one or more OpenSpliceAI model checkpoints (PyTorch
.ptor Keras.h5). The subcommand can average predictions across multiple models.
Output Files#
The primary output is a VCF file with added OpenSpliceAI annotations for each variant that passes filtering. Only SNVs and simple INDELs (REF or ALT is a single base) within genes are annotated. Variants in multiple genes have separate predictions for each gene. Each variant line in the annotated VCF contains a string in the INFO field with the format:
ALLELE|SYMBOL|DS_AG|DS_AL|DS_DG|DS_DL|DP_AG|DP_AL|DP_DG|DP_DL
Field |
Description |
|---|---|
ALLELE |
Alternate allele |
SYMBOL |
Gene symbol |
DS_AG |
Delta score (acceptor gain) |
DS_AL |
Delta score (acceptor loss) |
DS_DG |
Delta score (donor gain) |
DS_DL |
Delta score (donor loss) |
DP_AG |
Delta position (acceptor gain) |
DP_AL |
Delta position (acceptor loss) |
DP_DG |
Delta position (donor gain) |
DP_DL |
Delta position (donor loss) |
Delta Scores: Acceptor gain, acceptor loss, donor gain, and donor loss. The scores range from 0 to 1, and can be interpreted as the probability of the variant being splice-site-altering.
Delta Positions: Relative positions (±50 by default) of these maximum changes. Positive values are downstream, negative values are upstream.
For example, .. code-block:: text
A|MYGENE|0.27|0.00|0.09|0.02|3|-4|7|-2
This string shows: - Alternate allele is A - We are on MYGENE - The base positon 3 downstream of the variant has the highest acceptor gain score of 0.27 - The base position 4 upstream of the variant has the highest acceptor loss score of 0.00 (no loss) - The base position 7 downstream of the variant has the highest donor gain score of 0.09 - The base position 2 upstream of the variant has the highest donor loss score of 0.02
Delta Score Computation#
The “delta” score measures how much a mutation changes splice site predictions within a fixed window around the variant (default ±50 nucleotides, adjusted by -D parameter). For each variant, we compute reference predictions (\(d_{ref}\), \(a_{ref}\)) and alternative predictions (\(d_{alt}\), \(a_{alt}\)) for donor (\(d\)) and acceptor (\(a\)) channels:
where each maximum is taken over a window of 101 positions (±50) centered on the variant. The position of the maximum difference is recorded as the “delta position” (negative if upstream of the variant, positive if downstream).
Usage#
usage: openspliceai variant [-h] -R reference -A annotation [-I [input]] [-O [output]] [-D [distance]] [-M [mask]] [--model MODEL] [--flanking-size FLANKING_SIZE]
[--model-type {keras,pytorch}] [--precision PRECISION]
optional arguments:
-h, --help show this help message and exit
-R, --ref-genome reference
path to the reference genome fasta file
-A, --annotation annotation
"grch37" (GENCODE V24lift37 canonical annotation file in package), "grch38" (GENCODE V24 canonical annotation file in package), or path to a similar custom gene annotation file
-I, --input-vcf [input]
path to the input VCF file, defaults to standard in
-O, --output-vcf [output]
path to the output VCF file, defaults to standard out
-D, --distance [distance]
maximum distance between the variant and gained/lost splice site, defaults to 50
-M, --mask [mask] mask scores representing annotated acceptor/donor gain and unannotated acceptor/donor loss, defaults to 0
--model, -m MODEL Path to a SpliceAI model file, or path to a directory of SpliceAI models, or "SpliceAI" for the default model
--flanking-size, -f FLANKING_SIZE
Sum of flanking sequence lengths on each side of input (i.e. 40+40)
--model-type, -t {keras,pytorch}
Type of model file (keras or pytorch)
--precision, -p PRECISION
Number of decimal places to round the output scores
Examples#
Example: Discovering pathogenic human variants#
openspliceai variant \
--input-vcf input_variants.vcf \
--ref-genome GRCh38.fa \
--annotation grch38.txt \
--model /path/to/pytorch_models/ \
--model-type pytorch \
--flanking-size 400 \
--distance 100 \
--mask 1 \
--output-vcf annotated_variants.vcf
This command:
Loads the reference genome from
GRCh38.fa.Reads the gene annotation from
grch38.txt.Scans the directory
/path/to/pytorch_models/for PyTorch checkpoints, averaging predictions from all found models.Computes donor and acceptor delta scores within ±100 nucleotides of each variant.
Masks scores representing annotated acceptor/donor gain and unannotated acceptor/donor loss. (This is useful for novel/pathogenic variant discovery).
Writes a new VCF (
annotated_variants.vcf) with the masked annotations for the four delta scores and positions in theINFOfield.
Example: Annotating all variant in VCF#
openspliceai variant \
--input-vcf sample_variants.vcf \
--ref-genome GRCh37.fa \
--annotation grch37.txt \
--model /path/to/keras_models/ \
--model-type keras \
--flanking-size 10000 \
--output-vcf annotated_samples.vcf
--precision 3
This command:
Loads the reference genome from
GRCh37.fa.Reads the gene annotation from
grch37.txt.Scans the directory
/path/to/keras_models/for Keras checkpoints, averaging predictions from all found models.Computes donor and acceptor delta scores within ±50 nucleotides of each variant.
Writes a new VCF (
annotated_samples.vcf) with all annotations for the four delta scores and positions in theINFOfield (these are unmasked by default, so all scores are included). The scores are rounded to three decimal places.
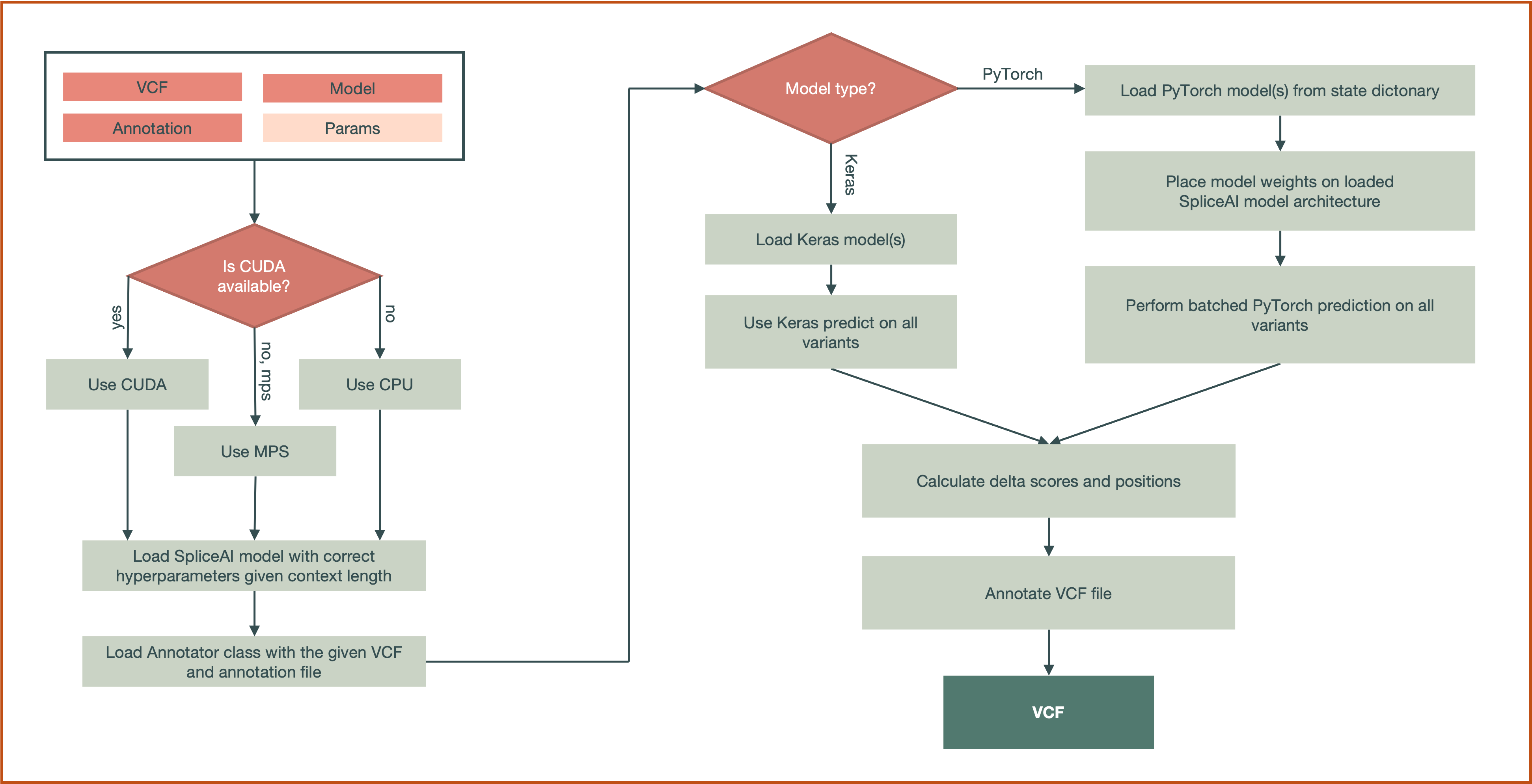
Processing Pipeline#
VCF Parsing/Filtering
For each variant, the subcommand checks if it lies within an annotated gene region. If it isn't, it will be filtered out.
Variants that are too close to the chromosome ends (<
flanking-size/ 2 bases on either side), have deletions of length > 2 *distance, or have reference alleles mismatching the FASTA are automatically skipped.
Reference & Mutant Sequence Extraction
A window of \(2 \times \text{dist_var} + 1\) around the variant is extracted from the FASTA (e.g., 101 nt for
--dist-var=50).For each alternative allele, the subcommand constructs a mutant sequence by substituting the variant base(s).
Model Loading & Prediction
The user may supply PyTorch (
.pt) or Keras (.h5) model checkpoints.If a directory is provided, predictions from each model are averaged.
Wild-type (reference) and mutant sequences are one-hot encoded and fed into the model(s) in evaluation mode.
Delta Score Calculation
VCF Annotation
The four delta scores and their positions (relative to the variant site) are appended to each variant’s INFO field.
The annotated VCF includes the annotations in the
INFOfield, following the string format described above.
Workflow#
Conclusion#
The variant subcommand enables fine-grained analysis of how single-nucleotide changes or small variants affect splice site usage. Essentially, it provides a convenient, post-hoc annotation step for variant effect prediction.