LiftOn's tutorial

Why LiftOn❓
Burgeoning number of genome assemblies: As of December 2023, there are 30,530 eukaryotic genomes, 567,228 prokaryotic genomes, and 66,429 viruses listed on NCBI (NCBI genome browser). However, genome annotation is lagging behind. As more high-quality assemblies are generated, we need an accurate lift-over tool to annotate them.
Improved protein-coding gene mapping: The popular Liftoff tool maps genes based on DNA alignments alone. Miniprot maps genes based on protein alignments but, without gene structure information, may not be as accurate on their own (See FAQ Common mistakes of Liftoff and miniprot). LiftOn combines both DNA-to-genome and protein-to-genome alignments and produces better gene mapping results! LiftOn improves upon the current released T2T-CHM13 annotation (JHU RefSeqv110 + Liftoff v5.1).
Improved distantly related species lift-over: A key limitation of DNA-based lift-over tools is that they do not perform well when the reference and target genomes have significantly diverged. With the help of protein alignments and the protein maximization algorithm, LiftOn improves the lift-over process between distantly related species. See "Mouse to Rat" and "Drosophila melanogaster to Drosophila erecta" result sections.
LiftOn is free, it's open source, it's easy to install, and it's in Python!
Who is it for❓
LiftOn is designed for researchers and bioinformaticians who are interested in genome annotation. It is an easy-to-install and easy-to-run command-line tool. Specifically, it is beneficial in the following scenarios:
If you have sequenced and assembled a new genome and require annotation, LiftOn provides an efficient solution for generating annotations for your genome.
LiftOn is an excellent tool for those looking to perform comparative genomics analysis. It facilitates the lifting over and comparison of gene contents between different genomes, aiding in understanding evolutionary relationships and functional genomics.
For researchers interested in using T2T-CHM13 annotations, try LiftOn! We have pre-generated the JHU_LiftOn_v1.0_chm13v2.0.gff3 file for your convenience.
What does LiftOn do❓
Let's first define the problem: Given a reference Genome \(R\), an Annotation \(R_A\), and a target Genome \(T\). The lift-over problem is defined as the process of changing the coordinates of Annotation \(R_A\) from Genome \(R\) to Genome \(T\), and generate a new annotation file Annotation \(T_A\). A simple illustration of the lift-over problem is shown in Figure 1.
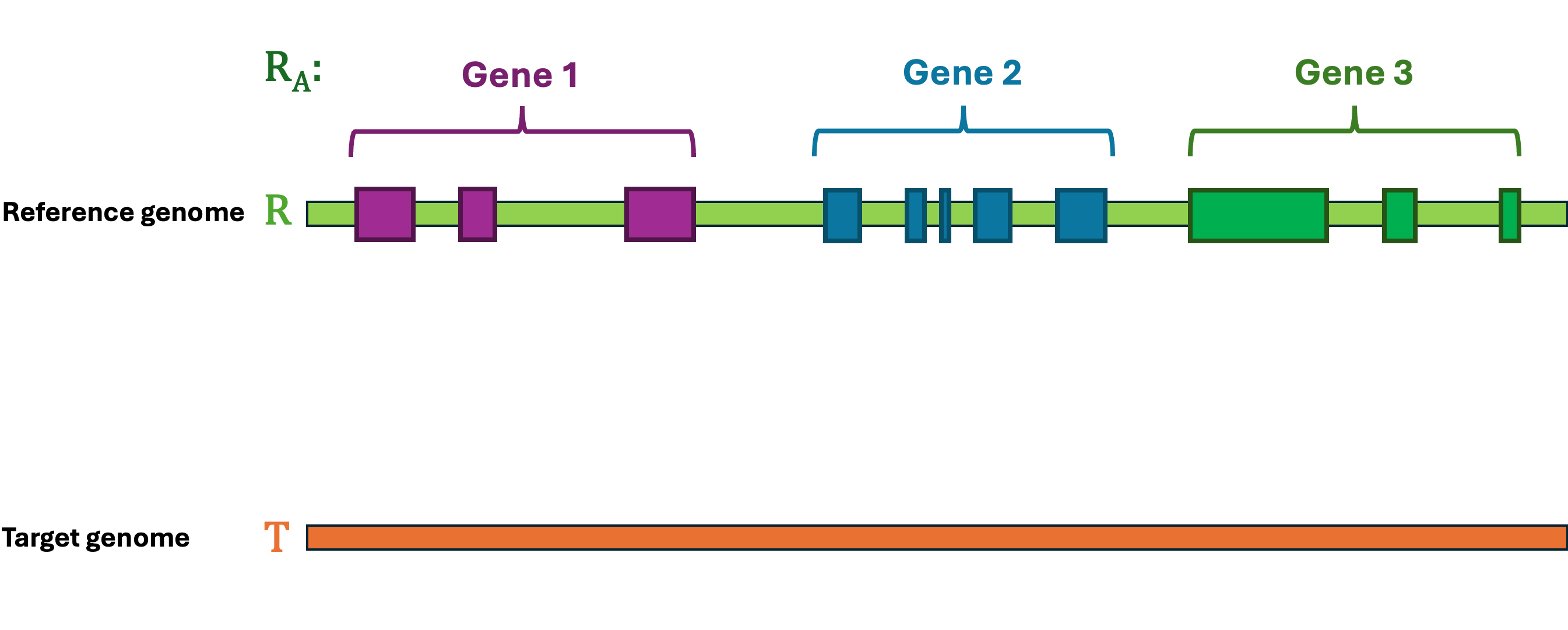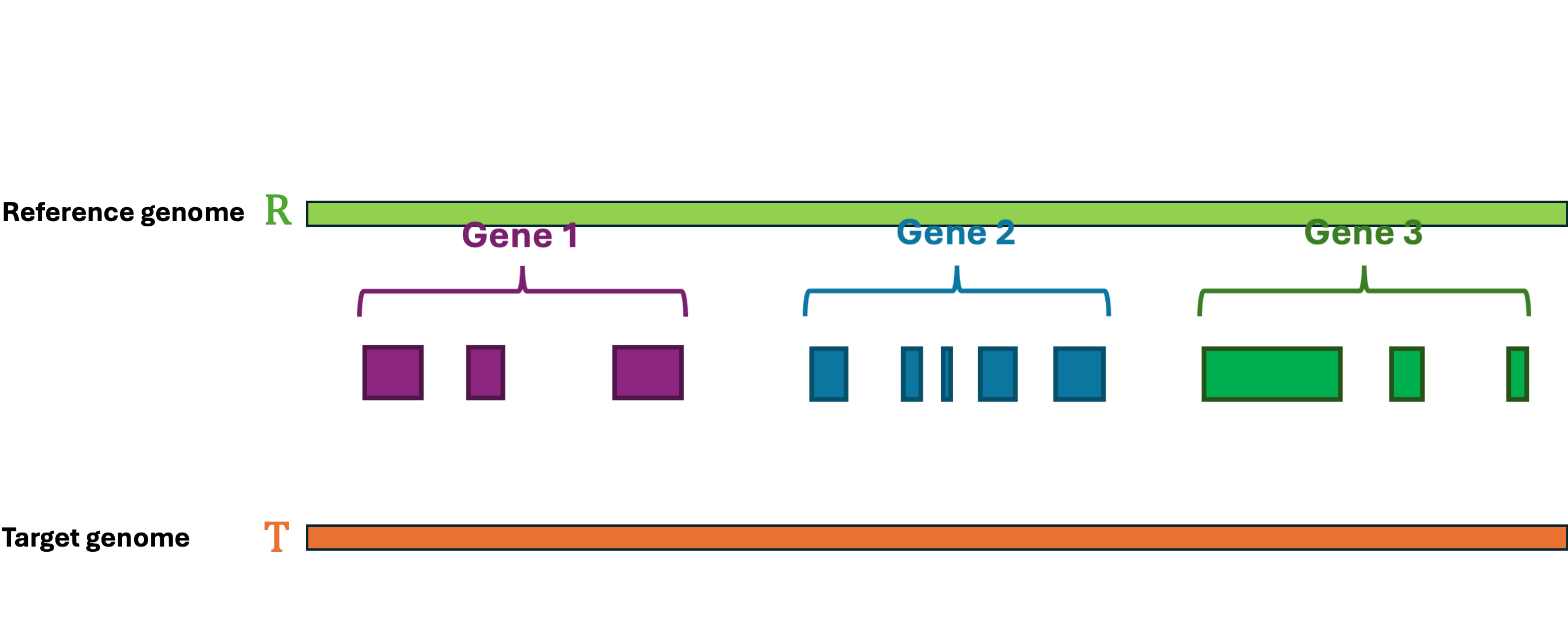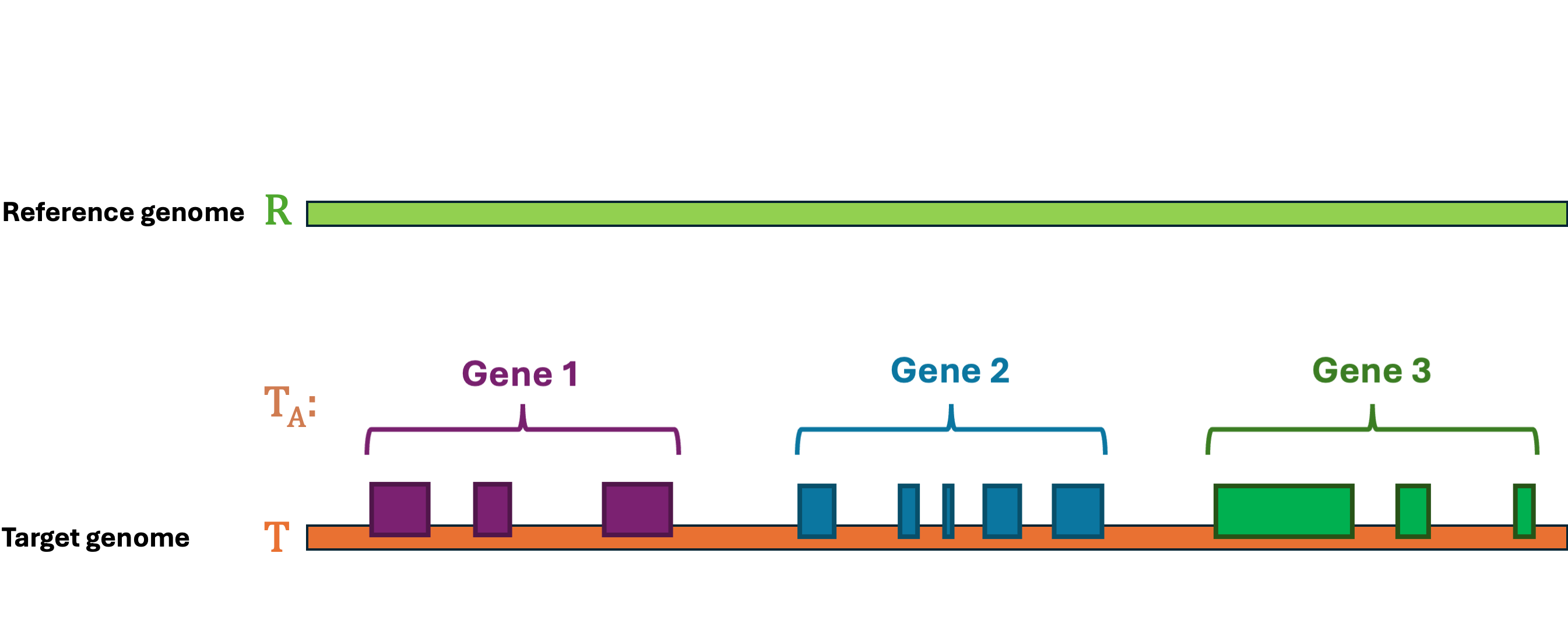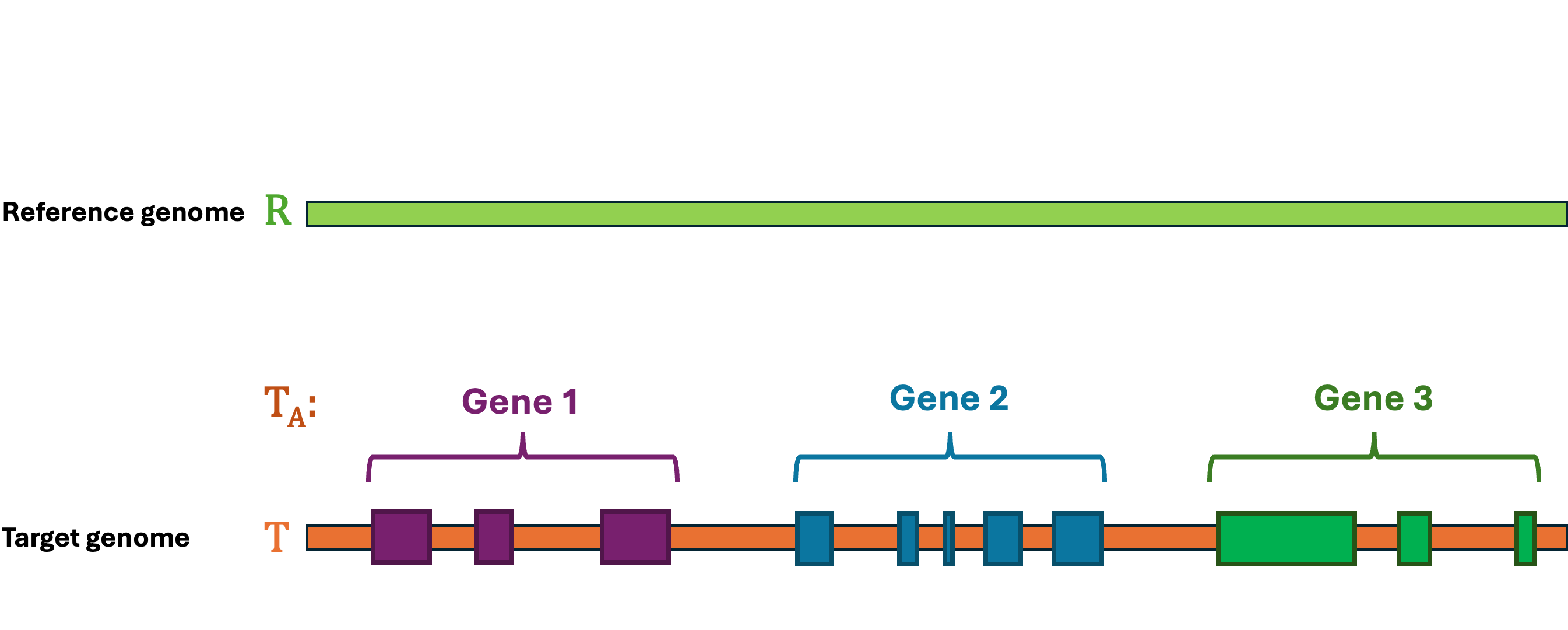
Figure 1 Illustration of the lift-over problem. The annotation file from the reference genome (top) is lifted over to the target genome (bottom).
LiftOn is the best tool to help you solve this problem! LiftOn employs a two-step protein maximization algorithm (PM algorithm).
The first module is the chaining algorithm. It starts by extracting protein sequences annotated by Liftoff and miniprot. LiftOn then aligns these sequences to full-length reference proteins. For each gene locus, LiftOn compares each section of the protein alignments from Liftoff and miniprot, chaining together the best combinations.
The second module is the open-reading frame search (ORF search) algorithm. In the case of truncated protein-coding transcripts, this algorithm examines alternative frames to identify the ORF that produces the longest match with the reference protein.
Inputs & Outputs
- Input:
Target Genome \(T\) in FASTA format.
Reference Genome \(R\) in FASTA format.
Reference Annotation \(R_A\) in GFF3 format.
- Output:
LiftOn annotation file, Annotation \(T_A\), in GFF3 format.
Protein sequence identities & mutation types.
Features with extra copies.
Unmapped features.
Cite us
Kuan-Hao Chao, Alan Mao, Steven L. Salzberg, and Mihaela Pertea. "Combining DNA and protein alignments to improve genome annotation with LiftOn." Genome Research 35.2, 311-325 (2025).
Alaina Shumate, and Steven L. Salzberg. "Liftoff: accurate mapping of gene annotations." Bioinformatics 37.12 (2021): 1639-1643.
User support
Please go through the documentation below first. If you have questions about using the package, a bug report, or a feature request, please use the GitHub issue tracker here:
https://github.com/Kuanhao-Chao/LiftOn/issues
Key contributors
LiftOn was designed and developed by Kuan-Hao Chao. This documentation was written by Kuan-Hao Chao and Alan Mao. The LiftOn logo was designed by Alan Mao.
Table of contents
Examples
Info
LiftOn's limitations
LiftOn's chaining algorithm currently only utilizes miniprot alignment results to fix the Liftoff annotation. However, it can be extended to chain together multiple DNA- and protein-based annotation files or aasembled RNA-Seq transcripts.
DNA- and protein-based methods still have some limitations. We are developing a module to merge the LiftOn annotation with the released curated annotations to generate better annotations.
The LiftOn chaining algorithm now does not support multi-threading. This functionality stands as our next targeted feature on the development horizon!