Benchmarks (v1.0.9)
LiftOn v1.0.9 was evaluated against the two single-method baselines it builds on — Liftoff (DNA / minimap2) and miniprot (protein) — and against the previous release LiftOn v1.0.8, in a four-way comparison spanning 17 whole genomes and 34 curated subsets across four evolutionary-distance tiers (same species → closely related → distant → very distant). Every annotation is re-scored with the same tool-neutral metric (Parasail global protein alignment; see the evaluation metrics page), so the four tools are compared on an even footing.
The full analysis — including the joint recall-vs-identity (apples-to-apples) breakdown and per-dataset tables — is available in the LiftOn v1.0.9 technical report and the peer-reviewed Genome Research paper.
Accuracy
Across all 17 whole genomes, LiftOn v1.0.9 produces protein annotations whose identity to the reference is at least as high as the best single-method baseline on every genome, and that lead over the single-method baselines grows with evolutionary distance (where the DNA lift and the protein lift each capture complementary signal). On Drosophila, for example, LiftOn v1.0.9 reaches a mean protein identity of 0.922, ahead of miniprot (0.913) and Liftoff (0.887), and slightly ahead of v1.0.8 (0.921).
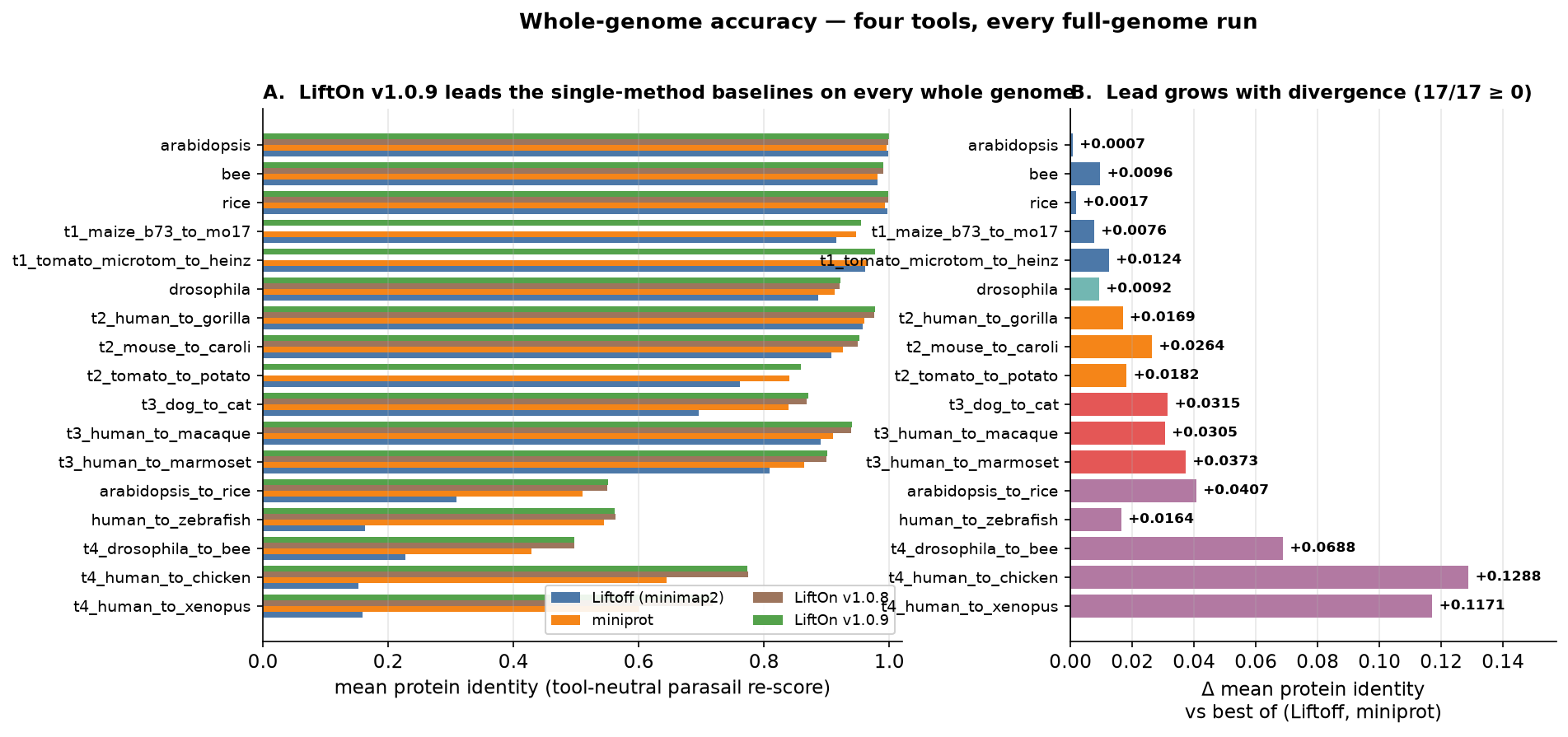
Figure 61 Whole-genome mean protein identity (tool-neutral Parasail re-score). A: LiftOn v1.0.9 (green) vs Liftoff, miniprot, and LiftOn v1.0.8 on every full genome. B: the gain over the best of the two single-method baselines, which widens with divergence.
Completeness & robustness
v1.0.9's crash-hardening lets full RefSeq / cross-species genomes that previously aborted run to completion. On the full Arabidopsis genome v1.0.8 crashed partway, recovering only ~28% of coding transcripts; v1.0.9 completes the run and recovers ~99.9%. Full rice improves the same way (77% → ~99.9%). All 17 whole-genome runs now finish.
The default gene-like lift additionally recovers reference feature types that
the previous gene-only lift dropped — on the full Arabidopsis genome that
adds +4,816 pseudogenes, +2,681 lnc_RNAs, plus tRNAs, ncRNAs, snoRNAs and
more.
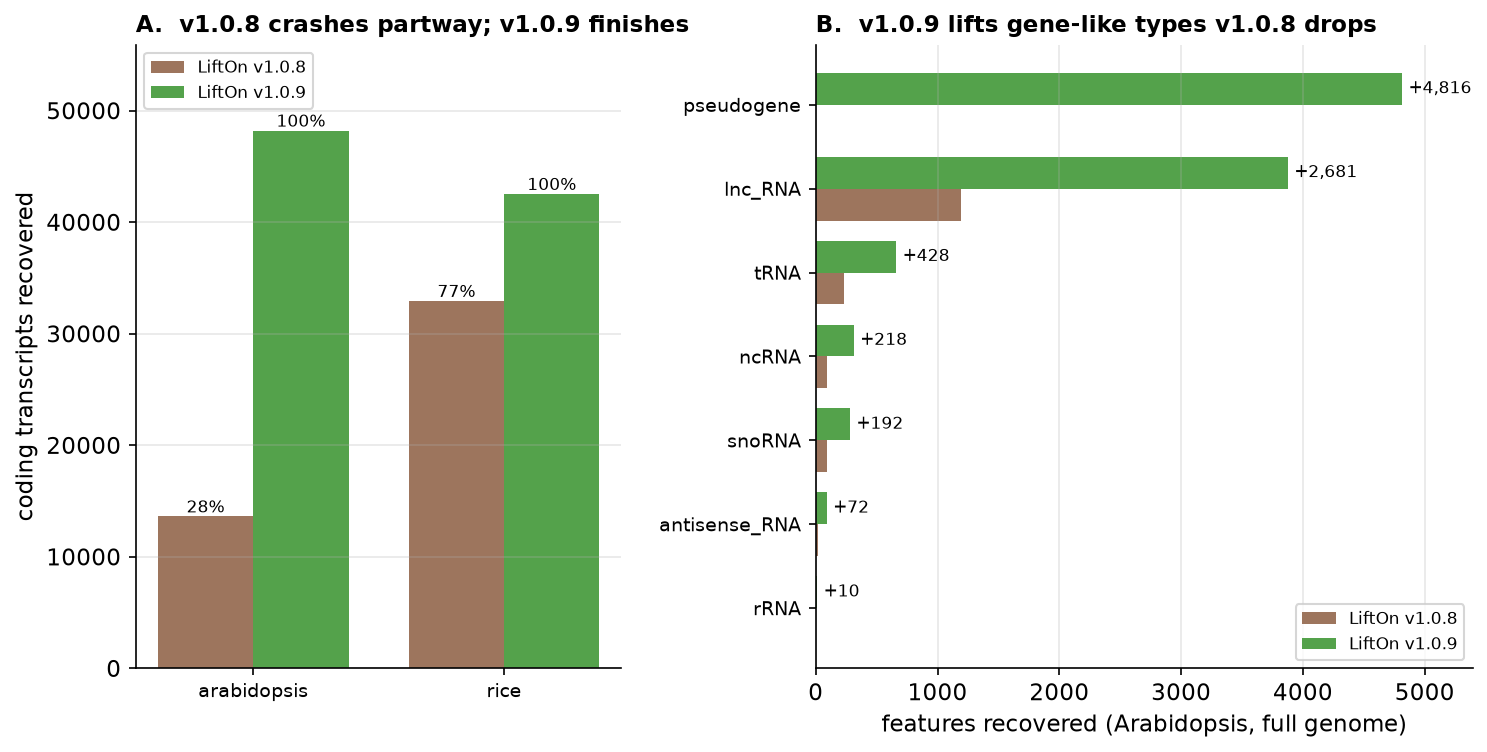
Figure 62 A: v1.0.8 crashes partway on full Arabidopsis / rice; v1.0.9 finishes (100% of the run). B: gene-like feature types recovered by v1.0.9 that v1.0.8 dropped (full Arabidopsis).
At the largest evolutionary distances, completeness becomes a deliberate precision–recall trade-off: miniprot's protein search recovers more loci (higher recall) at lower per-transcript identity, while LiftOn keeps Liftoff's DNA-anchored structure (higher identity) and fills genuinely-missing genes with the new miniprot-only rescue pass. The figure below shows where each tool sits; the technical report breaks this down further with a recall-weighted metric.
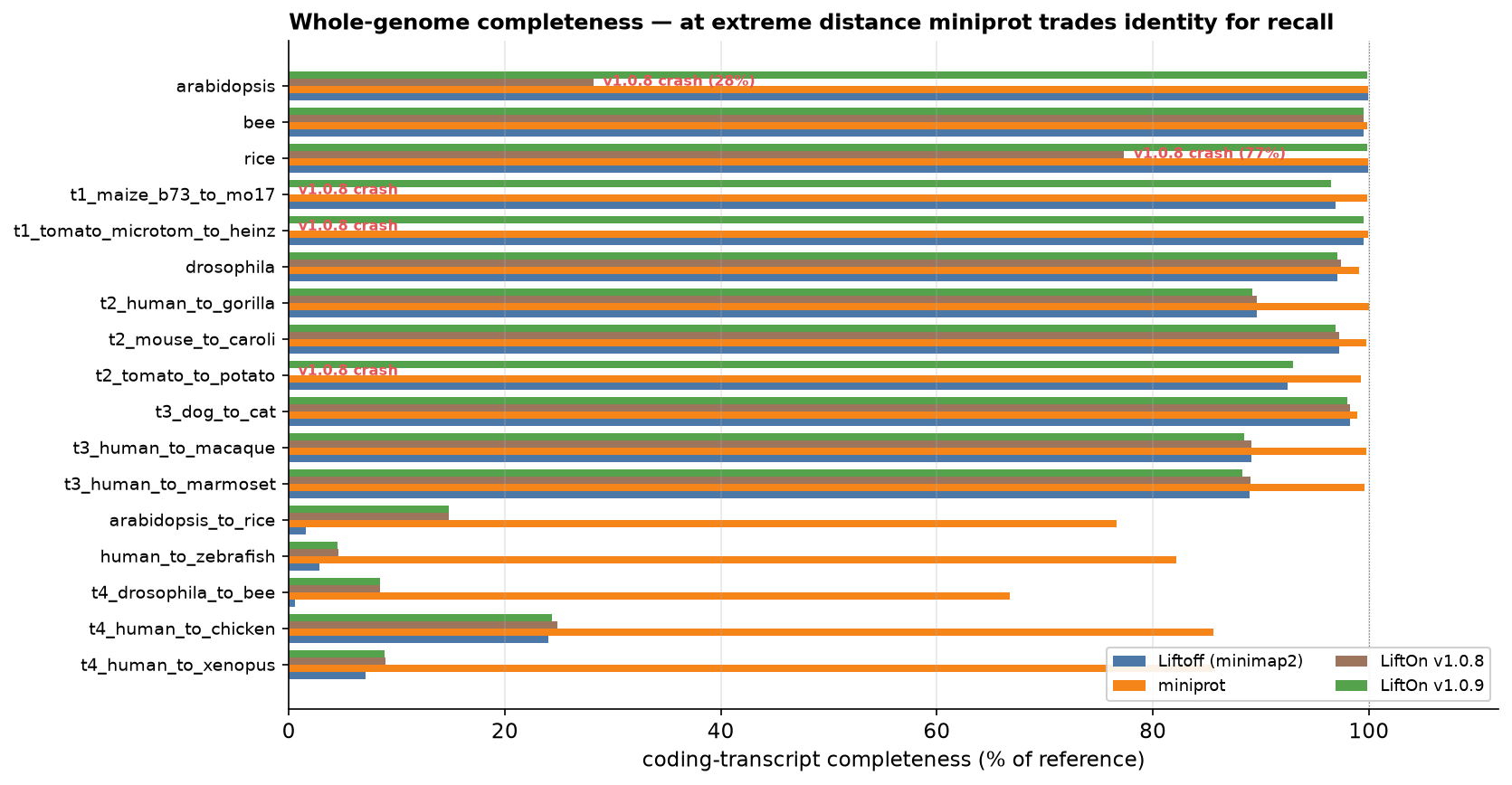
Figure 63 Coding-transcript completeness (% of reference) for all four tools. LiftOn v1.0.9 (green) completes every genome and tracks Liftoff closely; at the very distant tier miniprot (orange) trades identity for recall.
GFF3 validity
v1.0.9 makes the emitted annotation validate clean. Default exon/CDS
containment normalization and unique-exon-ID guarantees remove the
LiftOn-introduced containment and duplicate-ID errors that earlier versions
emitted (the reference annotations themselves do not have them). Measured with
the in-tree gff3-validate:
LiftOn v1.0.9 reports 0 validation errors on 9 of the 17 whole genomes (bee, Drosophila, human→zebrafish, human→gorilla, human→macaque, human→marmoset, human→chicken, human→xenopus, Drosophila→bee).
On all 17 genomes, v1.0.9 emits fewer errors than the DNA-only Liftoff, and it is dramatically cleaner than v1.0.8 on every genome both versions produced (e.g. dog→cat 159 → 15, Drosophila 150 → 0, all six human cross-species 107–150 → 0). (miniprot's count is pinned at the validator's 50-issues-per-check reporting cap, so it is not directly comparable on the high-error plant genomes.)
The residuals on organellar-gene-bearing plant genomes are reference-inherited, not LiftOn-introduced: the TAIR10 Arabidopsis reference itself carries 159 such errors — more than LiftOn's lifted output (49).
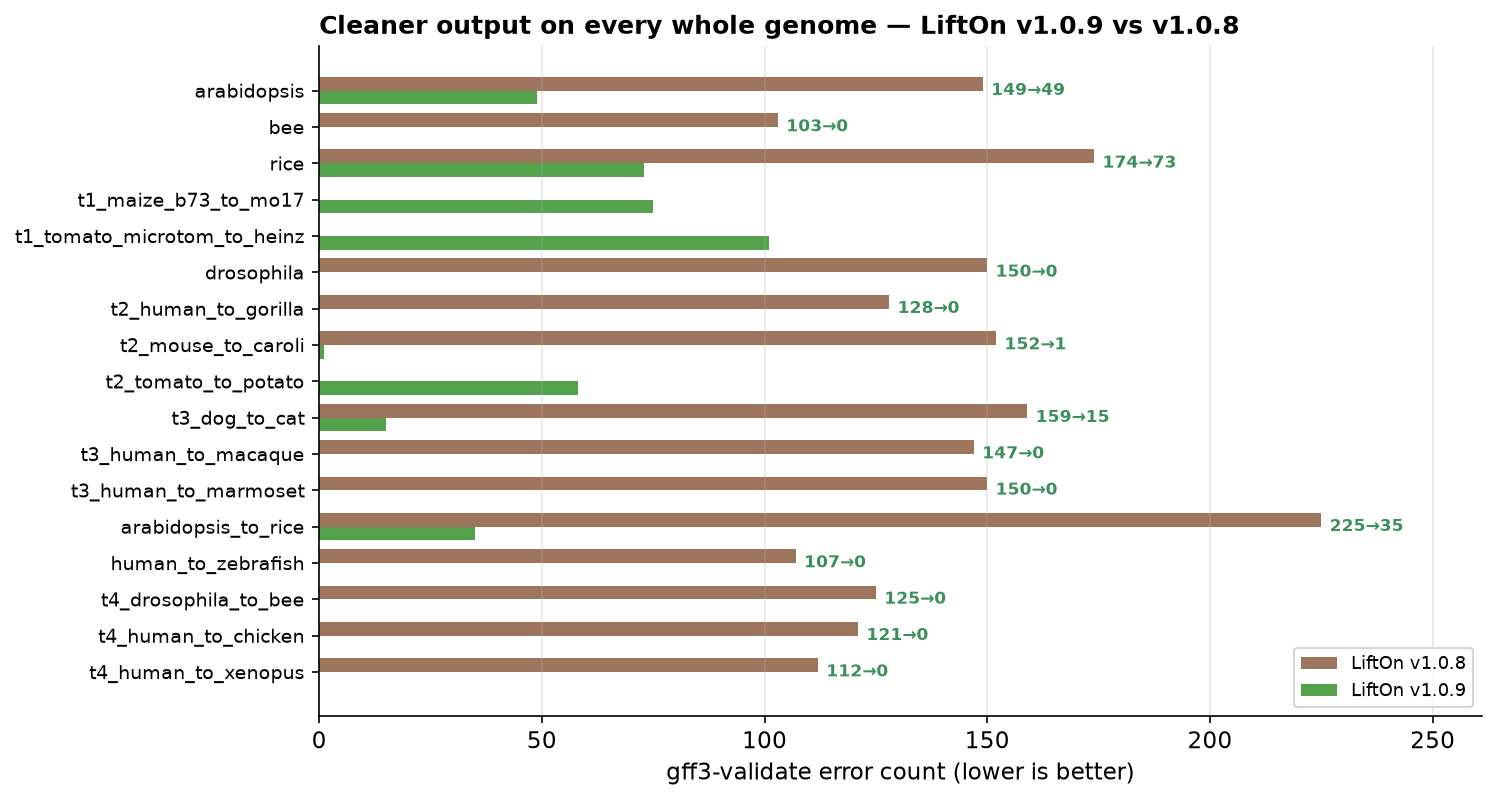
Figure 64 gff3-validate error count per whole genome, LiftOn v1.0.8 (brown) vs
v1.0.9 (green); lower is better.
Performance
v1.0.9 ships several byte-identical performance fast-paths — memory-bounded
windowed alignment for giant genes, concurrent Liftoff‖miniprot execution, a
fused per-locus thread pool, and a collapsed sequence-extraction query. These
change how LiftOn runs, never what it emits (pinned by a 24-cell
byte-identity test matrix). On a controlled single-thread arm with cached aligner
inputs (isolating LiftOn's own execution), v1.0.9 runs up to ~2.2× faster and
uses up to ~24× less peak memory than v1.0.8 — the giant-gene windowing is
what removes the multi-gigabyte memory peaks on mammalian / plant genomes.
Adding --threads N --locus-pipeline (also byte-identical) reduces wall-clock
further.
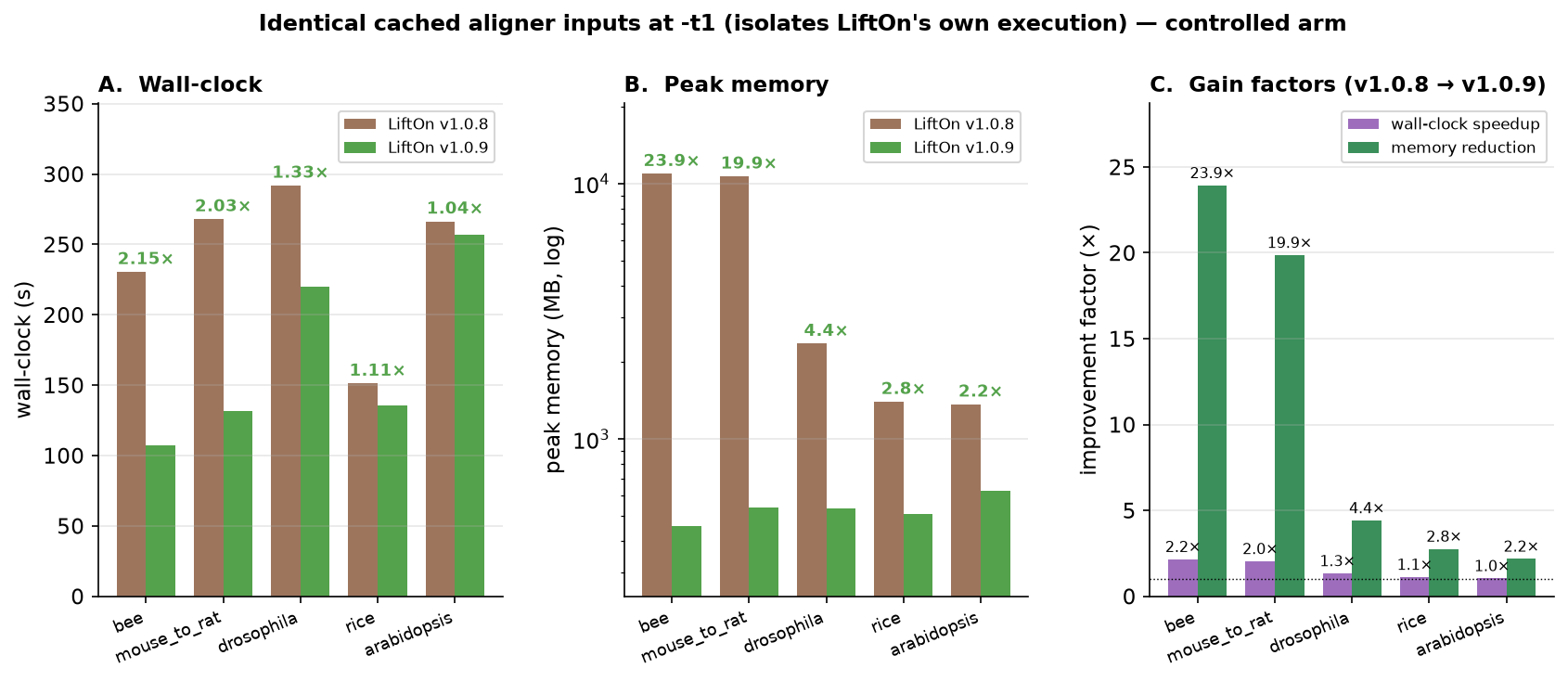
Figure 65 LiftOn v1.0.8 vs v1.0.9 on identical cached aligner inputs at -t 1. A:
wall-clock. B: peak memory (log scale). C: per-dataset gain factors
(wall-clock speedup and peak-memory reduction).
See the Changelog for the full list of v1.0.9 changes and their opt-out flags, and the User Manual for every command-line option.