Alignment file evaluation & cleanup (BAM)#
Introduction#
You are analyzing your RNA-Seq reads using a standard RNA-Seq pipeline. After performing quality control, you have utilized popular spliced aligners such as HISAT2 and STAR to align the reads to the genome. While inspecting the alignments in IGV, you have noticed that some reads are spliced and aligned across different gene loci or intergenic regions.
This raises concerns regarding the accuracy of these splice alignments and the authenticity of the reads originating from these locations. Additionally, you are curious if there is a systematic approach to validate these splice alignments. Unfortunately, there are currently no available tools that can assess the results generated by the aligners. Consequently,
Important
We propose running Splam as a new step in RNA-Seq analysis pipeline to score all splice junctions.
The power of Splam lies in its ability to filter splice alignments without relying on complex statistical rules. Instead, it utilizes our trained deep learning model to evaluate the DNA sequences surrounding the donor and acceptor sites. Splam learns the splice junction patterns. It identifies and removes splice alignments that exhibit low confidence splice junctions.
Splam offers solutions to several problems by:
Distinguishing high-quality spliced alignments from those with low confidence.
Providing a systematic approach to assess all splice alignments within an alignment file. Additionally, it eliminates erroneous splice alignments, thereby enhancing downstream transcriptome assembly.
Potentially aiding in understanding the biases inherent in splice aligners.
There are more potential usages to be explored!
Workflow overview#
Before diving into details of each step, here is an overview of the workflow. Figure 1 (a) is the workflow of running Splam in the standard RNA-Seq pipeline, and Figure 1 (b) is the workflow of spurious splice alignment removal.
Splam takes a sorted alignment file, extracts all splice junctions, and scores all of them. Furthermore, Splam cleans up the alignment file by removing spliced alignments with poor quality splice junctions, and outputs a new sorted alignment file.
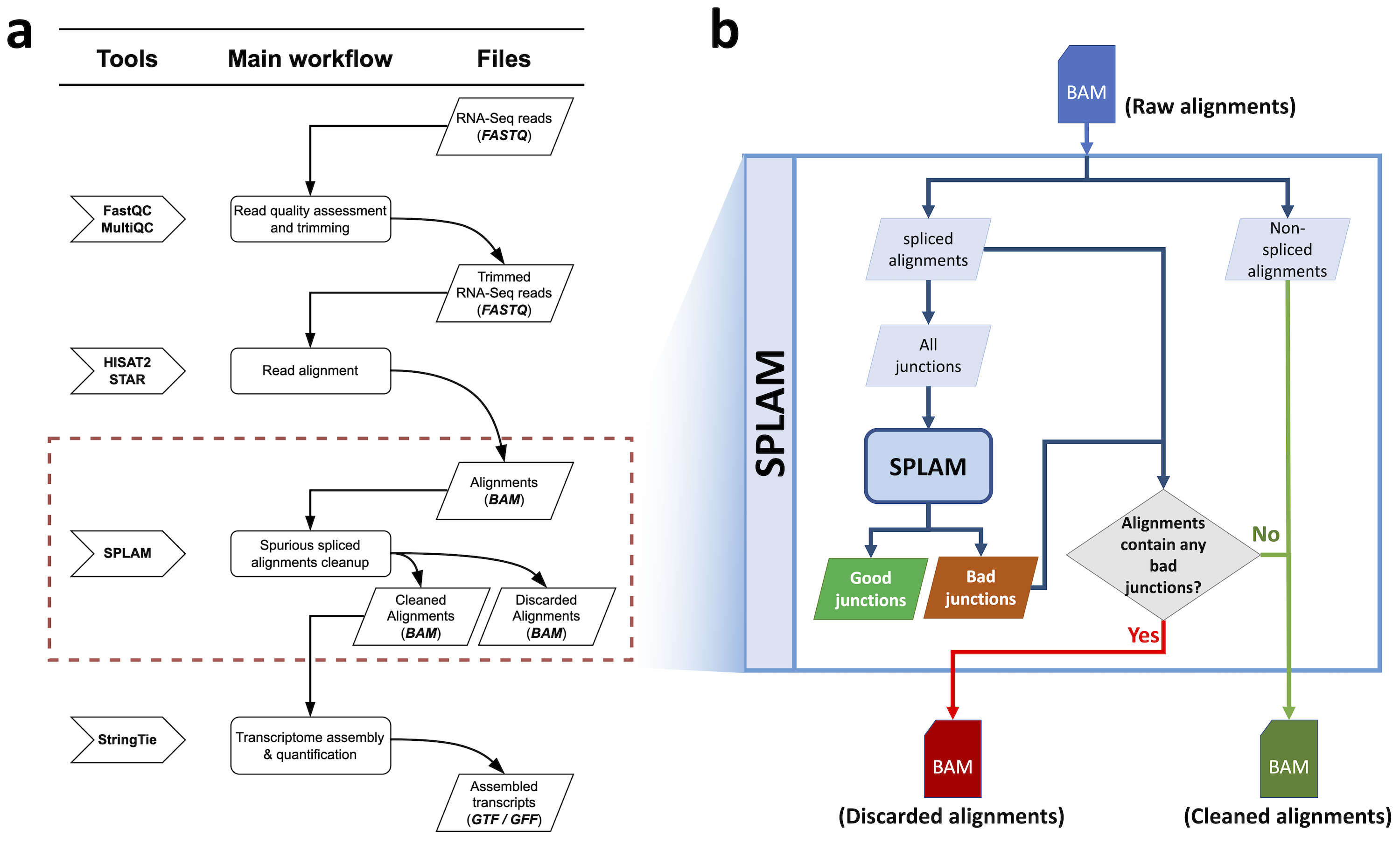
Figure 1 The Splam workflow for cleaning up spurious spliced alignments in an alignment file.#
Step 1: Preparing your input files#
The first step is to prepare three files for Splam analysis. In this tutorial, we will be using a toy dataset.
Input files
An alignment file in
BAMformat [example file: SRR1352129_chr9_sub.bam].A reference genome in
FASTAformat [example file: chr9_subset.fa].The Splam model, which you can find here: splam.pt
Step 2: Extracting splice junctions in your alignment file#
In this step, you take an alignment file (1) and run
splam extract -P SRR1352129_chr9_sub.bam -o tmp_out_alignment
The primary outputs for this step is a BED file containing the coordinates of each junction and some temporary files.
Splam iterates through the BAM file, extracts all splice junctions in alignments, and writes their coordinates into a BED file. By default, the BED is written into tmp_out/junction.bed. The BED file consists of six columns: CHROM, START, END, JUNC_NAME, INTRON_NUM, and STRAND. Here are a few entries from the BED file:
Output: junction.bed
1chr9 4849549 4860125 JUNC00000007 3 +
2chr9 5923308 5924658 JUNC00000008 6 -
3chr9 5924844 5929044 JUNC00000009 8 -
Note that in this command, we run with the argument -P / --paired. This argument should be selected based on the RNA sequencing read type. There are two types of RNA sequencing read types: single-read and paired-end sequencing. For a more detailed explanation, you can refer to this page.
By default, Splam processes alignments without pairing and bundling them. If your RNA-Seq sample is single-read, there is no need to set this argument. However, if your RNA-Seq sample is from paired-end sequencing, it is highly recommended to run Splam with the -P / --paired argument. Otherwise, if an alignment is removed, the flag of its mate will not be unpaired. It is worth noting that it takes longer to pair alignments in the BAM file, but it produces more accurate flags.
Here are some optional arguments:
Step 3: Scoring extracted splice junctions#
In this step, the goal is to score all the extracted splice junctions. To accomplish this, you will need 3 essential files. (1) The BED file that was generated in Step 2, (2) the reference genome (2) which shares coordinates with the junction BED file, and (3) the Splam model (3). Once you have these files in place, you can run the following command:
splam score -G chr9_subset.fa -m ../model/splam_script.pt -o tmp_out_alignment tmp_out_alignment/junction.bed
After this step, a new BED file is produced, featuring eight columns. Two extra columns, namely DONOR_SCORE and ACCEPTOR_SCORE, are appended to the file. It is worth noting that any unstranded introns are excluded from the output. (P.S. They might be from unstranded transcripts assembled by StringTie).
Output: junction_score.bed
1chr9 4849549 4860125 JUNC00000007 3 + 0.7723698 0.5370769
2chr9 5923308 5924658 JUNC00000008 6 - 0.9999831 0.9999958
3chr9 5924844 5929044 JUNC00000009 8 - 0.9999883 0.9999949
Here are the required arguments:
Here are some optional arguments:
Step 4: Cleaning up your alignment file#
After scoring every splice junction in your alignment file, the final step of this analysis is to remove alignments with low-quality splice junctions and update 'NH' tags and flags for multi-mapped reads. You can pass the directory path to Splam using the clean mode, which will output a new cleaned and sorted BAM file. The implementation of this step utilizes the core functions of samtools sort and samtools merge. If you want to run this step with multiple threads, you can set the -@ / --threads argument accordingly.
splam clean -P -o tmp_out_alignment -@ 5
Output: cleaned.bam
The output file of this step is a sorted Splam-cleaned BAM file. You can replace the original BAM file with this cleaned BAM file to do the transcript assembly, quantification, and all other downstream analyses!
Splam score threshold suggestion
For cleaning up BAM alignment files, we advise using a more lenient score threshold of 0.1. That being said, Splam is a decisive model and performs quite consistently across a wide range of thresholds, so a score threshold between 0.1 to 0.9 would work well.
Here are some optional arguments:
Step 5: IGV visualization#
Here is an example of the EHMT1 gene locus on chromosome 9 visualized in IGV. This protein-coding gene is located on the forward strand; however, we have observed that the splice aligner generates several splice alignments on the reverse strand.
In Figure 2, the first three tracks display the coverage, splice junction, and alignment information from the original alignment file of the SRR1352129 sample. The fourth, fifth, and sixth tracks show the coverage, splice junction, and alignment data obtained from the cleaned alignment file of the SRR1352129 sample, which was generated using Splam. Many of the spliced alignments on the reverse strand of EHMT1 have splice junctions with low Splam scores and were consequently removed. The Splam removal procedure results in a more refined gene locus and enhances the transcriptome assembly. The final track represents the RefSeq annotations of the EHMT1 gene.
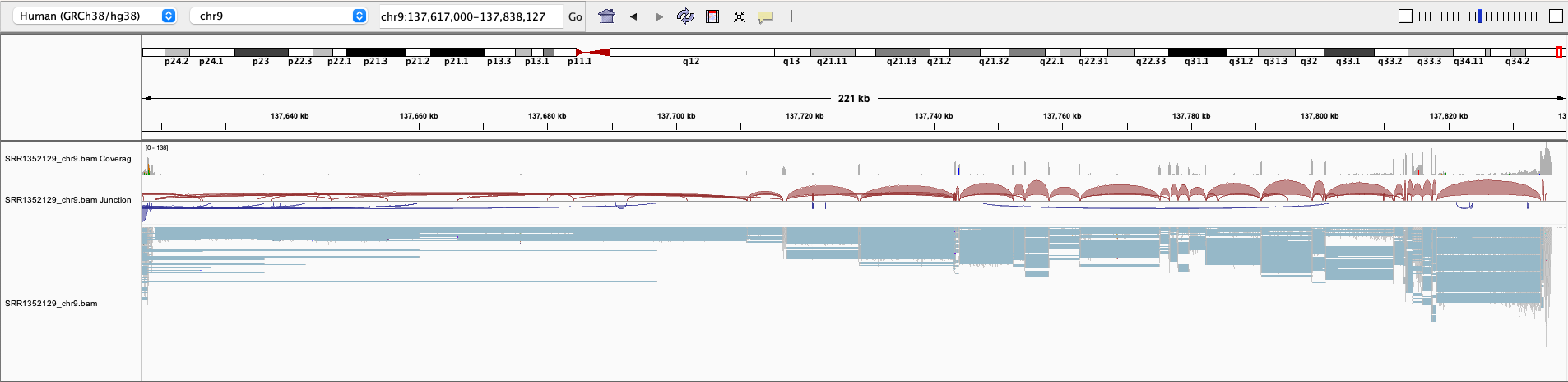
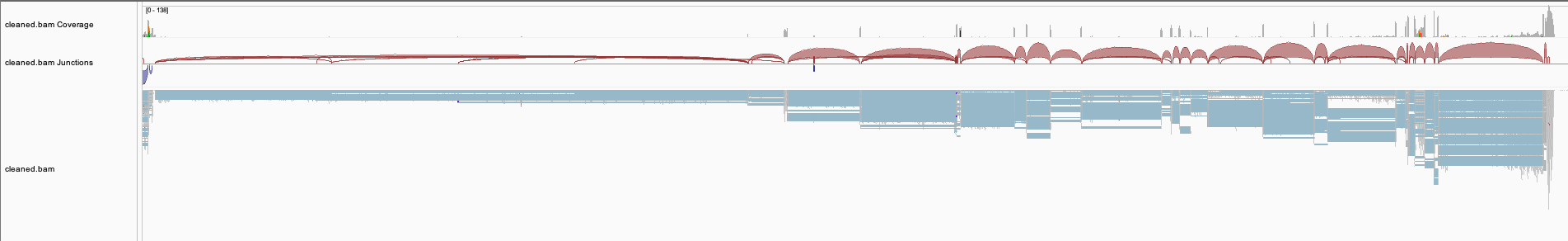
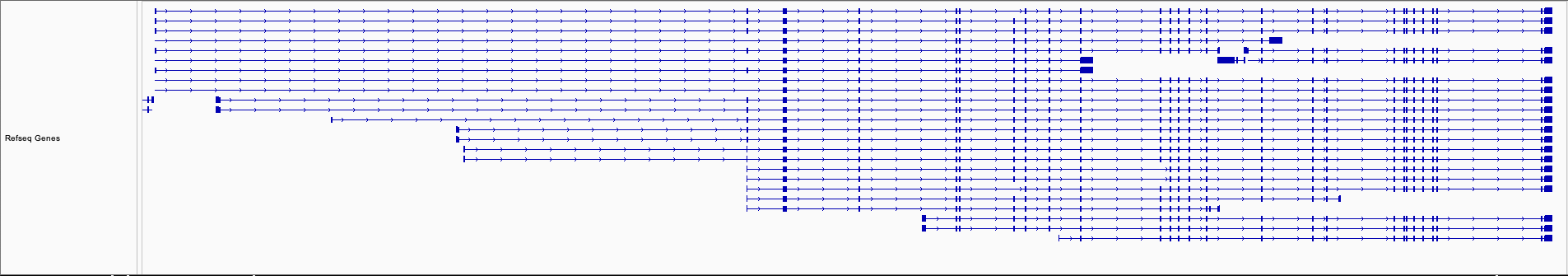
Figure 2 An example of a BAM file before and after Splam cleanup.#
Important
Splam exclusively employs the strand information to extract the reverse complement of DNA sequences for splice junctions when necessary. When it comes to scoring splice junctions, Splam relies solely on the DNA sequence information.
In the above example, Splam can distinguish that the majority of splice junctions aligned on the opposite strand of the EHMT1 gene locus are of poor quality. This final score is drawn by simply examining the DNA sequence!
Step 6: Assembling alignments into transcripts#
We ran Stringtie to assemble the original alignment BAM file and the Splam-cleaned alignment BAM file. Subsequently, we loaded both sets of assembled transcripts along with the RefSeq annotation into IGV (Figure 3). Upon observation, we noted that at the EHMT1 gene locus, there was originally one transcript assembled on the opposite strand of this gene, which will no longer be assembled after applying Splam's cleaning process, and the 3' end of the transcripts become more accurate!
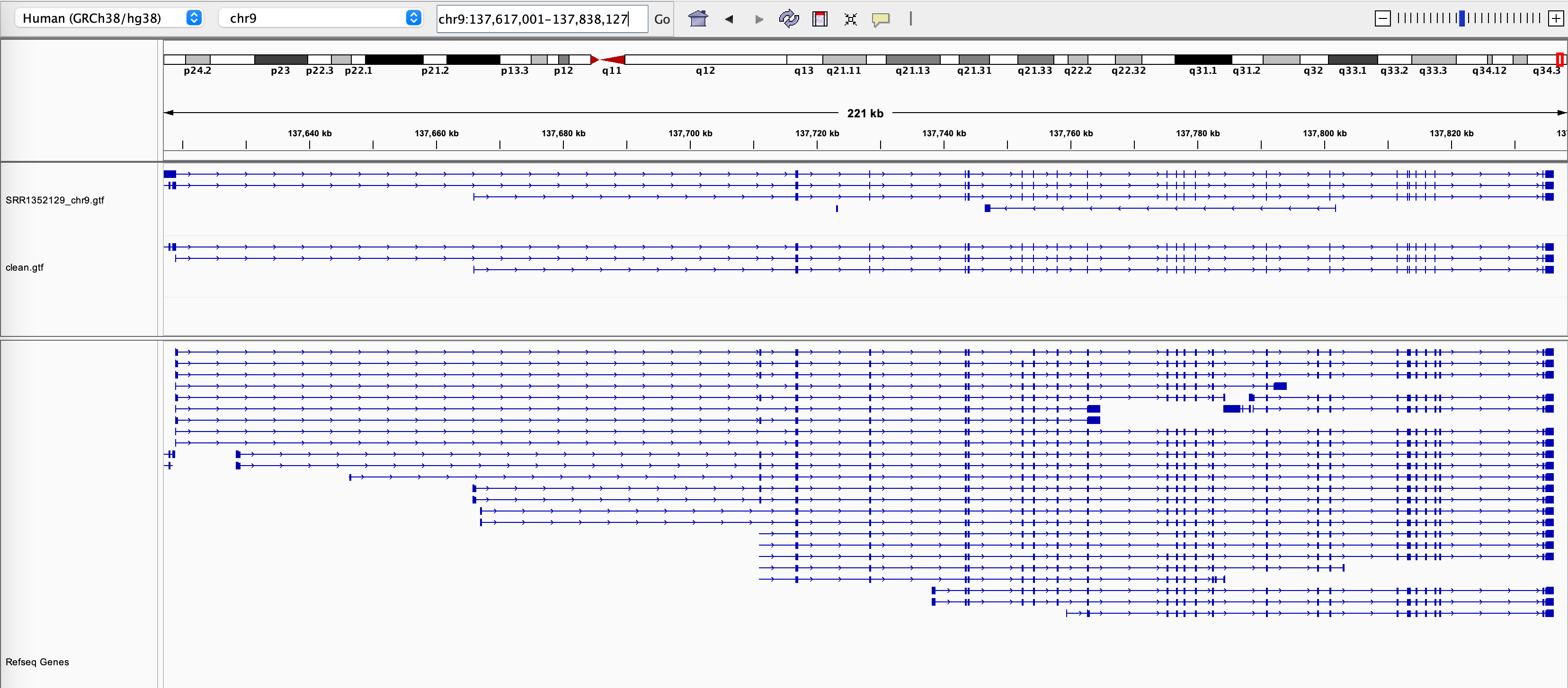
Figure 3 The assembly results of the original alignment file and the Splam-cleaned alignment file.#
See also
StringTie to learn more about the transcriptome assembly
What's next?#
Congratulations! You have finished this tutorial.
See also
Behind the scenes to understand how Splam is designed and trained
Q & A ... to check out some common questions