Splam's tutorial#

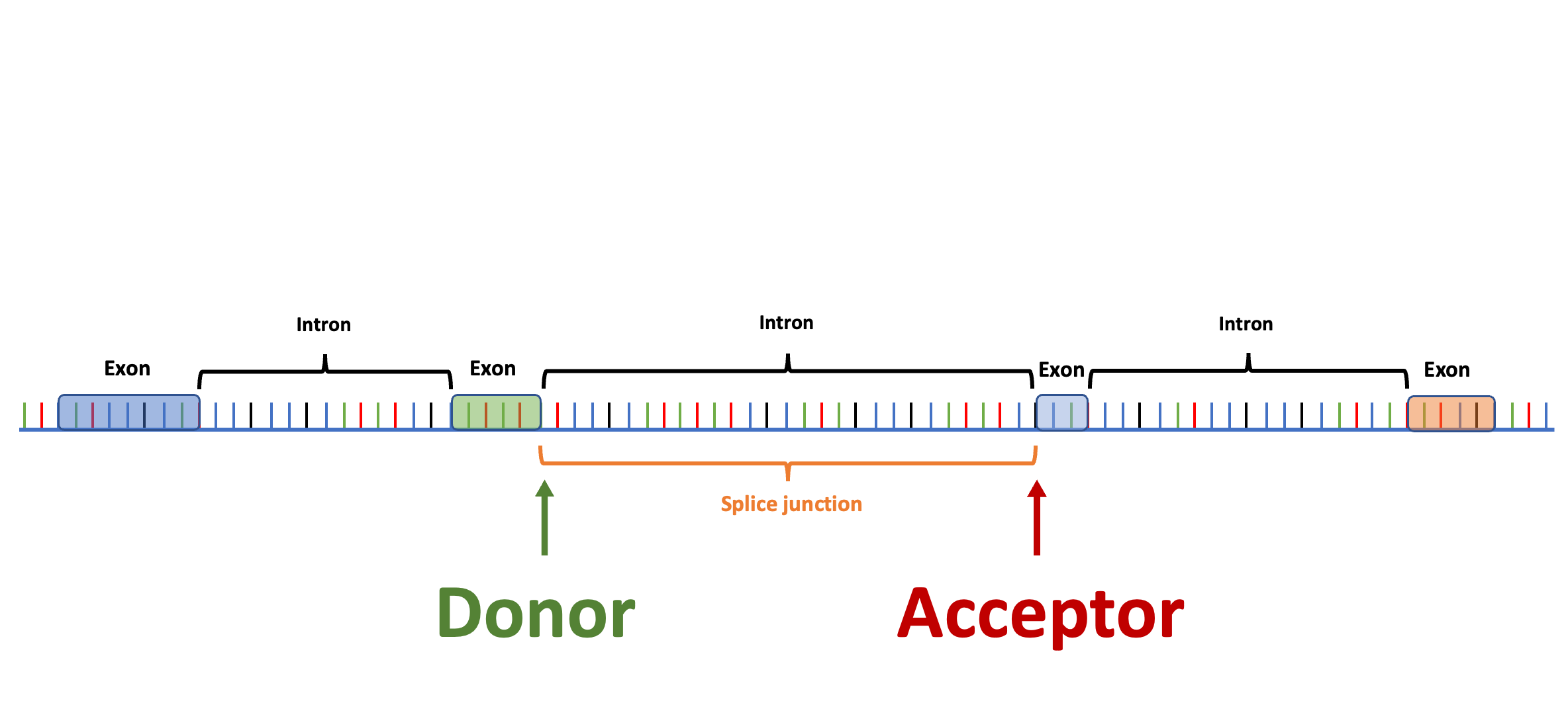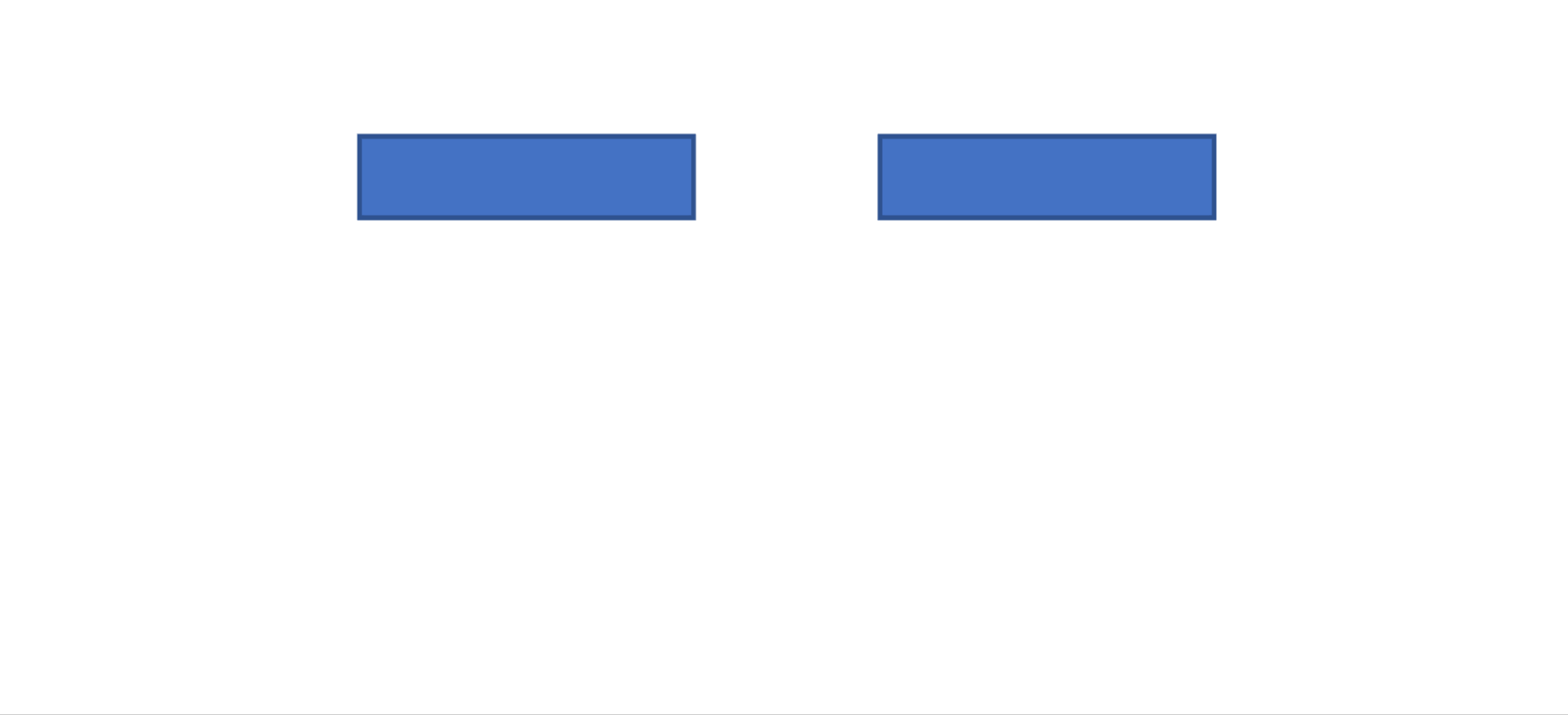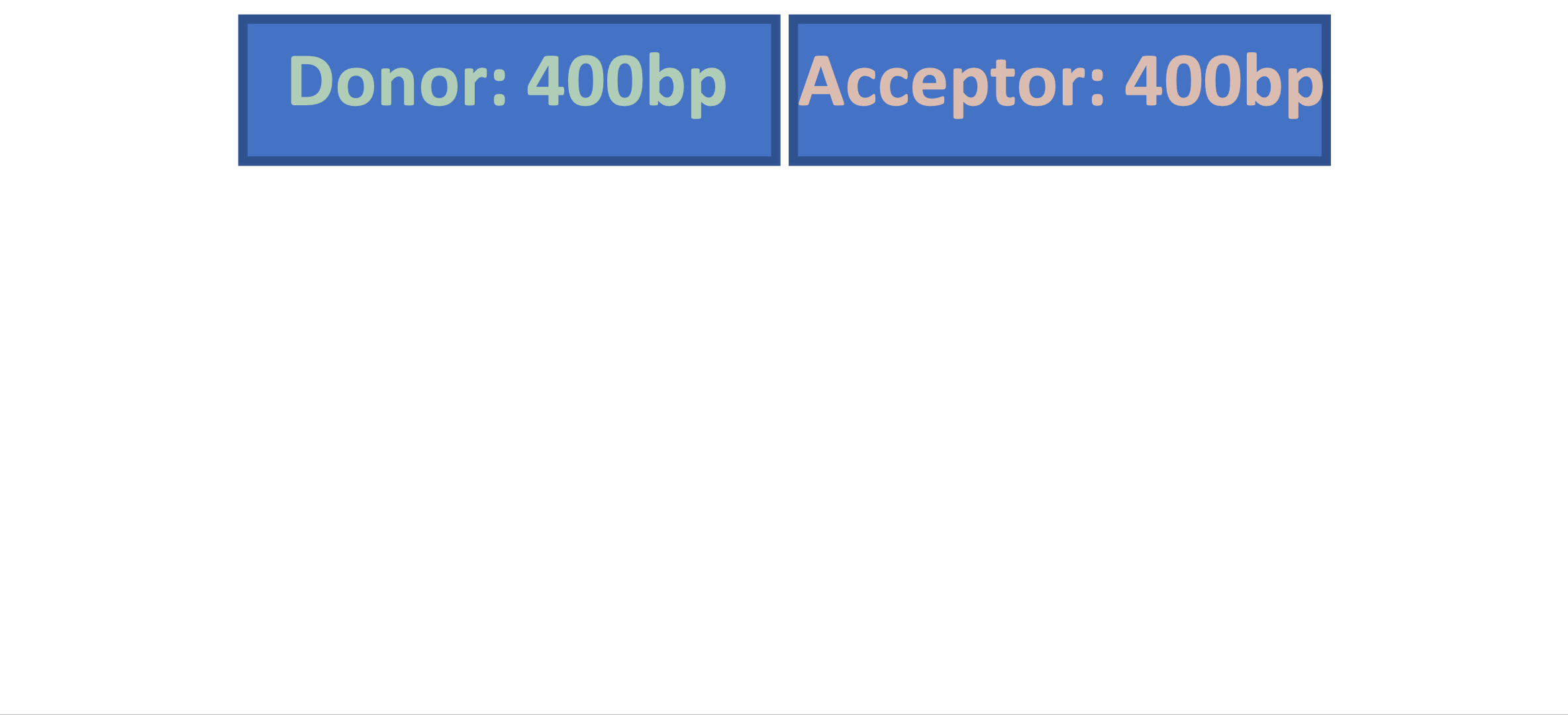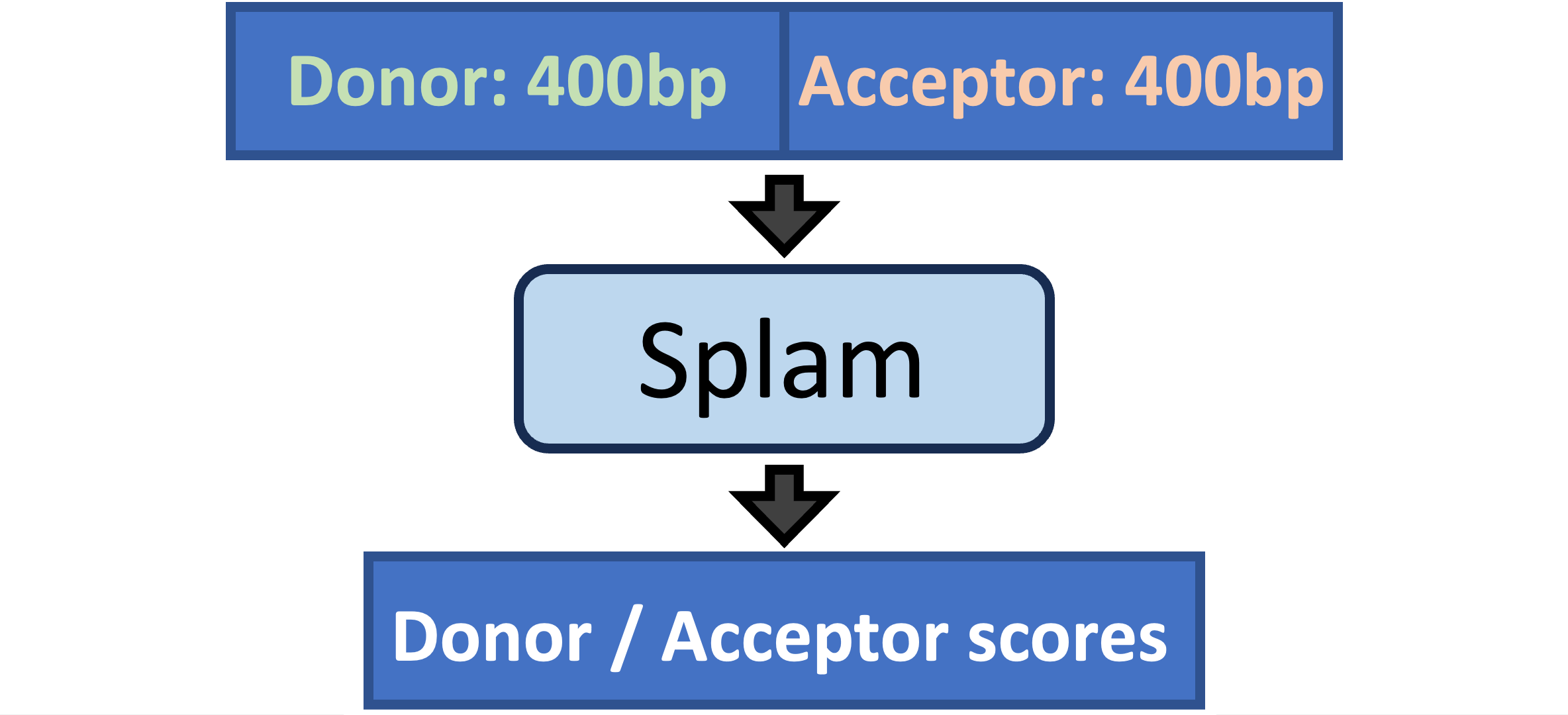
Splam is a splice site predictor utilizing a deep residual convolutional neural network for fast and accurate evaluation of splice junctions solely based on 400nt DNA sequences around donor and acceptor sites.
Why Splam❓#
We need a tool to evaluate splice junctions & spliced alignments. Thousands of RNA-Seq datasets are generated every day, but there are no tools available for cleaning up spurious spliced alignments in these data. Splam addresses this problem!
Splam-cleaned alignments lead to improved transcript assembly, which, in turn, may enhance all downstream RNA-Seq analyses, including transcript quantification, differential gene expression analysis, and more!
Who is it for❓#
If you are (1) doing RNA-Seq data analysis or (2) seeking a trustworthy tool to evaluate splice junctions (introns), then Splam is the tool that you are looking for!
What does Splam do❓#
There are two main use case scenarios:
Improving your alignment file. Splam evaluates the quality of spliced alignments and removes those containing spurious splice junctions. This significantly enhances the quality of downstream transcriptomes [Link].
Evaluating the quality of introns in your annotation file or assembled transcripts [Link].
Splam is free, it's open source, it's a lightweight deep learning model, and it's in Python!
Main features#
Biologically inspired training process: Splam was trained on combined donor and acceptor pairs, emulating the behavior of the spliceosome, with a specific emphasis on a narrow window of 400 base pairs surrounding each splice site. This approach is inspired by the understanding that the splicing process predominantly relies on signals within this specific region.
Generalization to non-human species: Splam was trained exclusively using human splice junctions; however, we have demonstrated that it performs well on chimpanzee, mouse, and even the flowering plant Arabidopsis thaliana.
Python & C++ integration: We have taken care of all the engineering work for you! Splam is easy to install and runs efficiently due to its underlying C++ implementation. You can install and run Splam with just one simple command.
Run Splam in three steps: With just three lines of code, you can obtain a new alignment file that is cleaned and sorted.
Pytorch implementation: Splam is implemented and trained using the popular and reliable PyTorch framework.
What Splam doesn't do#
Splam does not have the feature to scan through the genome and score every potential splice site. Some splice site prediction tools take a DNA sequence and predict the splice sites within it, such as SpliceAI. However, SpliceAI was only trained on a single canonical transcript for each protein-coding gene while disregarding alternative isoforms. Splam takes a different approach, focusing on predicting at the "splice junction level" rather than the "transcript level." Splam was trained on a large collection of human splices sites taken from both "canonical" and alternative isoforms.
Cite us#
Kuan-Hao Chao, Jakob M. Heinz, Celine Hoh, Alan Mao, Alaina Shumate, Mihaela Pertea, and Steven L. Salzberg. "Splam: a deep-learning-based splice site predictor that improves spliced alignments." Genome Biology 25, 243 (2024).
User support#
Please go through the documentation below first. You can find common questions and answers in the FAQ section. If you have questions about using the package, a bug report, or a feature request, please use the GitHub issue tracker here:
https://github.com/Kuanhao-Chao/splam/issues
Key contributors#
Splam deep residual convolutional neural network was trained using the PyTorch framework by Kuan-Hao Chao. Kuan-Hao Chao also implemented the package that applies Splam to evaluate annotation files and clean up alignment files. Alan Mao benchmarked Splam on non-human species. This documentation was written by Kuan-Hao Chao and Alan Mao.