Behind the scenes#
Data curation#
Data plays a critically important role in deep learning. The model performance largely depends on the quality, quantity, and relevance of the data used for training.
To understand how Splam works, you first need to understand how the training data was generated. For curating input splice sites, our approach differs from the state-of-the-art tool, SpliceAI, which was trained on splice sites labeled on canonical transcripts only. Our approach focuses on a smaller region that encompasses the donor and acceptor sites.
We extract a 400bp sequence centered around the splice sites, shifting the focus from learning the splicing pattern at the transcript level to the splice site level. This strategy enables localized prediction of splicing events, mitigating biases associated with canonical transcripts and eliminating the need for larger window sizes encompassing the entire intron and 10,000bp flanking sequences. In summary, each data point in our Splam model consists of a donor-acceptor pair with a 400bp window size for each, resulting in a total of 800bp.
Model input & output dimension#
The input to the model is a 800bp one-hot-encoded DNA sequence in the dimension of 4 x 800. The output is a 3 x 800 array, with every column representing a base pair, and each row reflecting its probability of being a donor site, acceptor site, or neither (as shown in Figure 6). The three scores sum up to 1. For the development of Splam, we used the Pytorch version 1.13.0 framework.
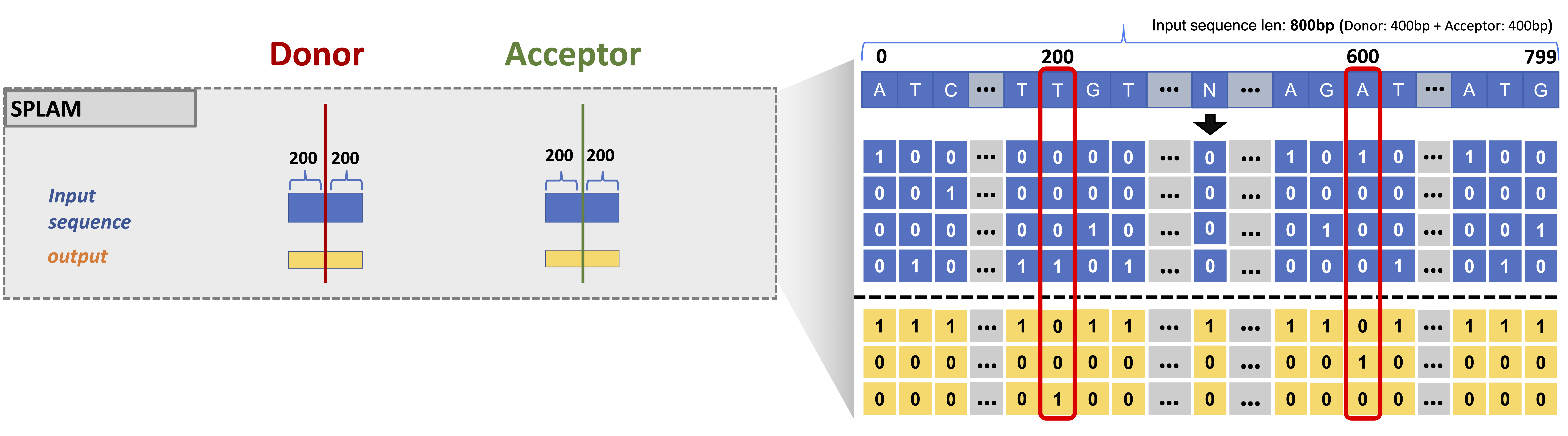
Figure 6 Splam takes 400bp regions around a donor and acceptor site, resulting in a total of 800bp. The input sequences are one-hot encoded into vectors, where \(A\) is \([1, 0, 0, 0]\), \(C\) is \([0, 1, 0, 0]\), \(G\) is \([0, 0, 1, 0]\), \(T\) is \([0, 0, 0, 1]\) and \(N\) is \([0, 0, 0, 0]\),. For the labels, a donor site is represented as \([0, 0, 1]\), an acceptor site as \([0, 1, 0]\), and an non-junction site as \([1, 0, 0]\).#
Positive data#
We filtered the junctions by extracting only those that were supported by more than 100 alignments in the Tiebrush merged BAM file. Furthermore, we used RefSeq GRCh38 patch 14 annotations to extract all protein-coding genes and intersected all 100-alignment-supported splice sites with the protein-coding splice sites observed in the RefSeq database.
The resulting splice sites were then categorized as Positive-MANE (blue in Figure 7) if they were in the Ensembl GRCh38 MANE release 1.0 file, and Positive-ALT (green in Figure 7) if they were only in RefSeq GRCh38 patch 14 but not in MANE.
This approach provides expression evidence to support splice junctions and thus eliminates transcription noise, increasing our confidence in the extracted donor and acceptor sites. Overall, we identified 180,195 splice sites in Positive MANE and 85,908 splice sites in Positive-ALT
Negative data#
Curating high-quality negative splice junctions is a challenging task due to incomplete annotations, unknown functions, and potential transcriptional noise. Labeling unannotated junctions as negatives without caution may misclassify some spliced junctions. Various methods can be used to generate negative splice junctions, such as selecting random dinucleotide pairs or random GT-AG pairs from the genome. However, these artificial junctions may differ significantly from true splice sites, leading to the learning of non-critical patterns by the model, resulting in low sensitivity and precision.
To address this issue in training Splam, two novel approaches were adopted for generating challenging negative splice junctions. (1) The first approach involved selecting random GT-AG pairs on the opposite strand of protein-coding gene loci. Since overlapping genes are rare in eukaryotes, it is unlikely to have another transcribed gene on the opposite strand of a protein-coding gene. This resulted in 4,467,910 splice junctions referred to as Negative-Random (red in Figure 7).
To further increase the difficulty of negative junctions, (2) only splice junctions with 1-alignment support on the opposite strand of protein-coding gene loci were chosen. These junctions are likely to be transcriptional noise or alignment artifacts and should be eliminated, making them more suitable for Splam training. This approach generated 2,486,305 splice junctions referred to as Negative-1 (orange in Figure 7). Accurately curating these challenging negative splice junctions allowed for effective training of Splam.
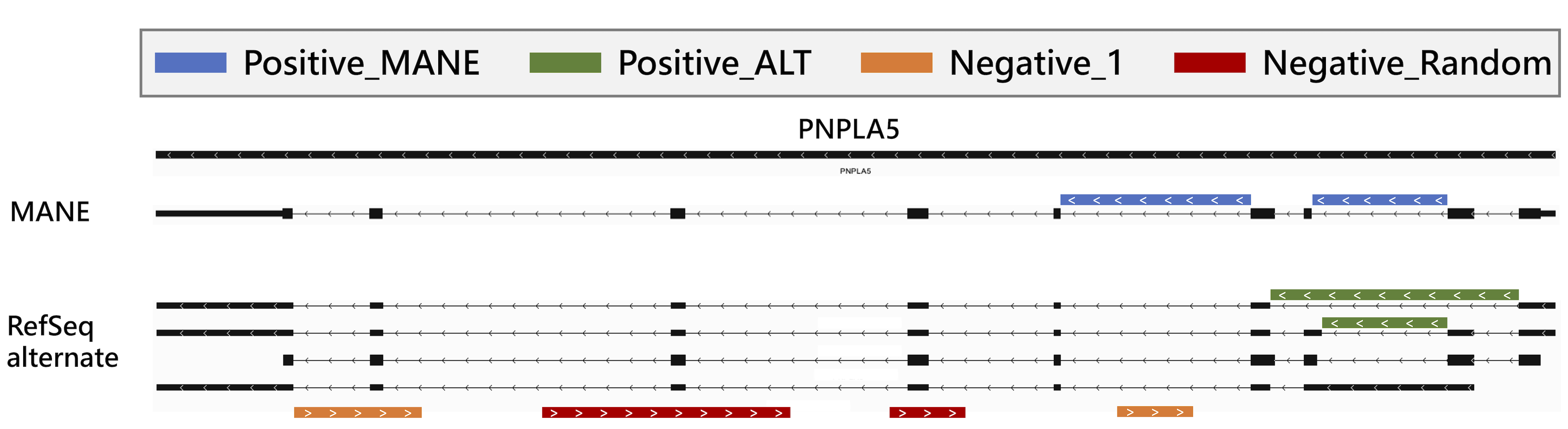
Figure 7 Illustration of the 4 types of splice sites used for training and testing Splam: Positive-MANE, Positive-ALT, Negative-1, and Negative-Random. Positive-MANE sites (blue) are selected from the MANE database and supported by at least 100 alignments, while Positive-ALT (green) are present in the RefSeq database but missing from MANE, and also supported by at least 100 alignments. Negative-1 sites (orange) occur on the opposite strand of a known gene and are supported by only 1 alignment, and Negative Random sites (red) are random GT-AG pairs on the opposite strand that do not overlap with any known splice sites and have no alignment support.#
Model architecture#
Splam utilizes a deep dilated residual convolutional neural network (CNN) that incorporates grouped convolution layers within the residual units.
Residual unit#
Splam architecture consists of 20 residual units, each containing two convolutional layers. The model uses a grouped convolution approach with a parameter called group set to 4. The hyperparameters of Splam include F (number of filters), W (window size), D (dilation rate), and G (groups), which are shown as (F, W, D, G) in Figure 8 (b). The concept of grouped convolution, which allows for memory saving with minimal accuracy loss, is inspired by the ResNext model.
Each convolutional layer in the residual unit follows a batch normalization and a rectified linear unit (ReLU) Figure 8 (b), and the input of the unit is residually connected to its output. He et al. [1] introduced residual units to address the issue of training accuracy degradation in deep learning. The inclusion of shortcut connections enables successful training of deeper models using simple stochastic gradient descent (SGD) with backpropagation, leading to improved accuracy as the depth increases.
Residual group#
A group of four residual units forms a bigger residual group, and 20 RUs are clustered into five residual groups. Residual groups are stacked such that the output of the i th residual group is connected to the i+1 th residual group. Furthermore, the output of each residual group undergoes a convolutional layer with the parameters (64, 1, 1), and then being added to all the other outputs of residual groups (residual connections colored in red), which then is passed into the last convolutional layer in (3, 1, 1) and a softmax layer. F is set to 64 for all convolutional layers, and for each residual group, W is set to 11, 11, 11, 21, and 21, and D is set to 1, 5, 10, 15, and 20 in residual groups in sequence. G is by default is 1 for all convolutional layers, but set to 4 in the residual units. We visualized the architecture of Splam in Figure 8. For each nucleotide position, its total neighboring span of the Splam model is
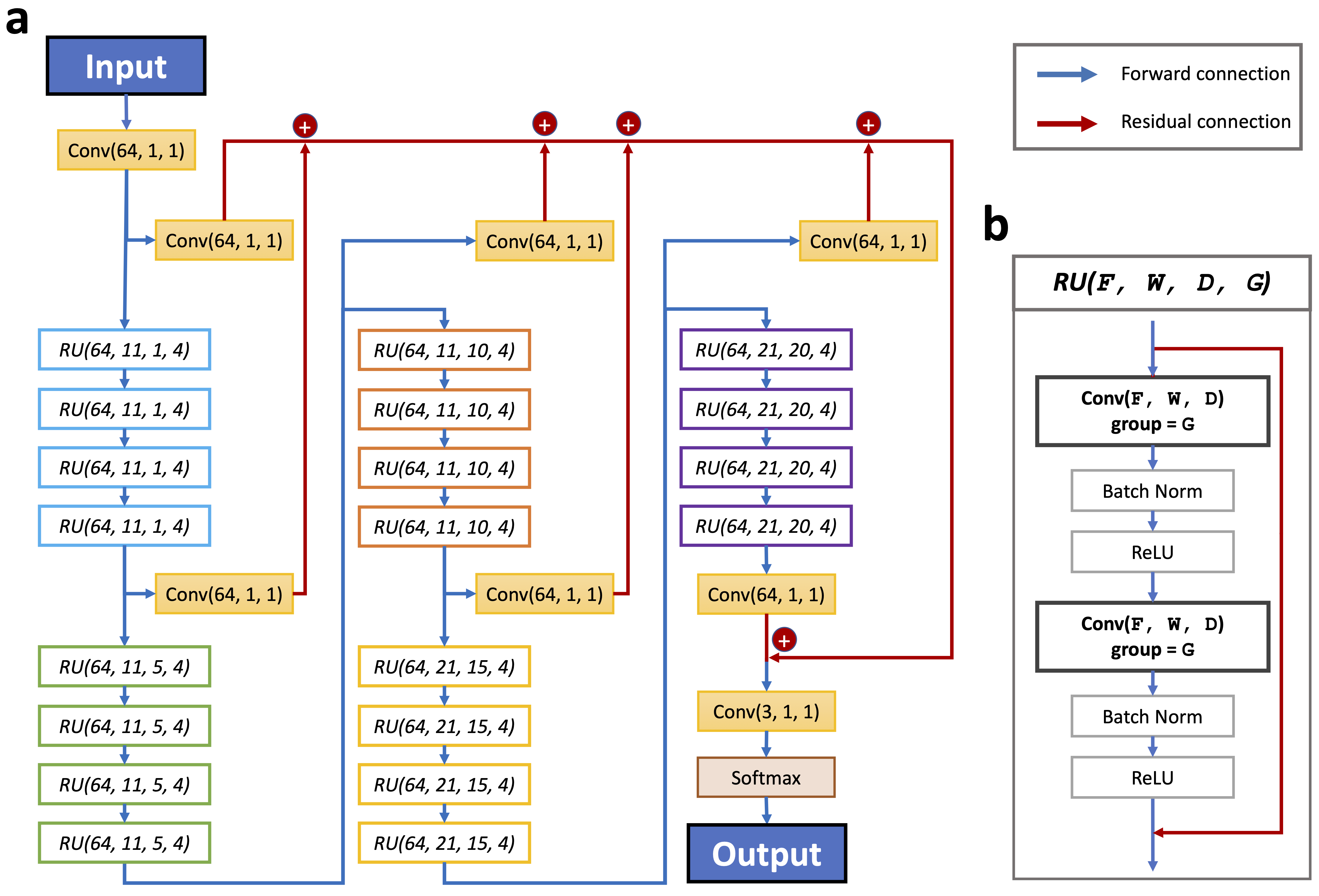
Figure 8 The model architecture of the Splam deep residual convolutional neural network.#
Splam training & testing#
After curating the gold standard dataset, we divided all splice junctions into two datasets: one for model training and the other for testing. For model training, we utilized all the splice sites on the main chromosomes, except chromosomes 1 and 9. For model testing, we used the splice sites on the held-out chromosomes 1 and 9, with the splice sites in paralogs removed.
Hyperparameters#
To train Splam, we used a batch size of 100 and trained it for 15 epochs. We employed the AdamW optimizer with the default learning rate of 0.03. A 1000-step warmup was utilized, with the learning rate increasing linearly from 0 to 0.03. The learning rate then decreased following the values of the cosine function between 0.03 to 0 (Figure 9).

Figure 9 The learning rate for each Splam update during training#
Loss function#
We further improved Splam's performance by changing the loss function. Instead of using the commonly used cross entropy (Eq. 1), we replaced it with focal loss (Eq. 2) [2].
Focal loss puts more emphasis on the challenging data points where Splam is more likely to make incorrect predictions and penalized these data points by an additional \((1-P)^{\gamma}`\) scale, where \(\gamma = 2\) and \(P\) is the probability of each class. This scale quantifies the degree of inaccuracy in predictions, instead of simply binary misclassifications that cross entropy applies.
Reference#
Kaiming He, Xiangyu Zhang, Shaoqing Ren, and Jian Sun. Deep residual learning for image recognition. In Proceedings of the IEEE conference on computer vision and pattern recognition, 770–778. 2016.
Tsung-Yi Lin, Priya Goyal, Ross Girshick, Kaiming He, and Piotr Dollár. Focal loss for dense object detection. In Proceedings of the IEEE international conference on computer vision, 2980–2988. 2017.