Q & A ...#
Q: What makes Splam different from SpliceAI?
Splam and SpliceAI are both frameworks used for predicting splice junctions in DNA sequences, but they have some key differences.
Input constraints:
Splam: Follows the design principle of using biologically realistic input constraints. It uses a window limited to 200 base pairs on each side of the donor and acceptor sites, totaling 800 base pairs. Furthermore, we pair each donor and acceptor as follows
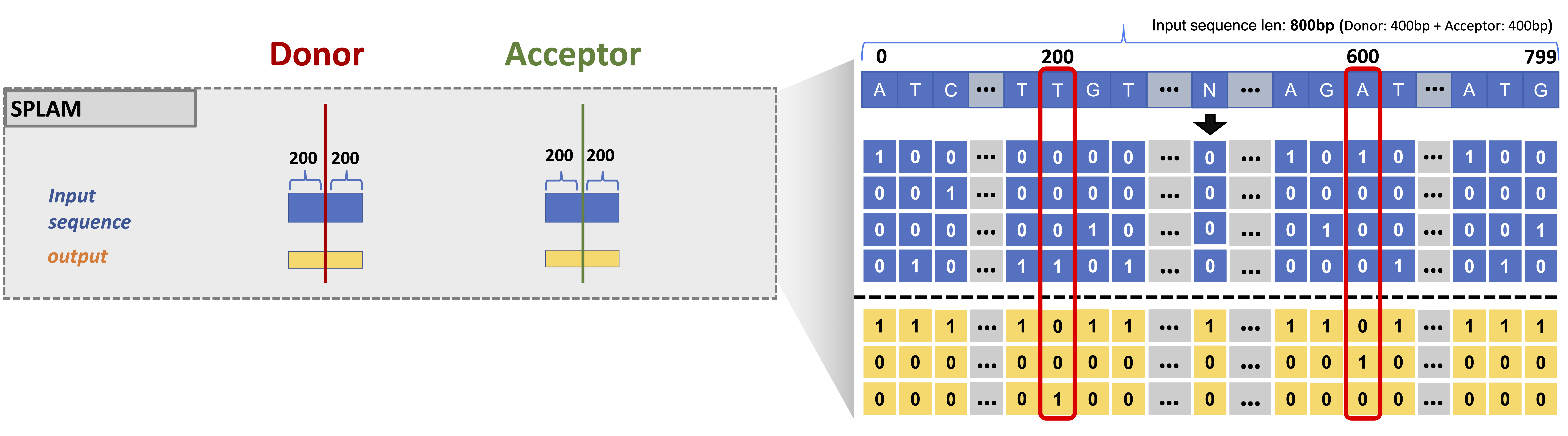
SpliceAI: The previous state-of-the-art CNN-based system, SpliceAI, relies on a window of 10,000 base pairs flanking each splice site to obtain maximal accuracy. However, this window size is much larger than what the splicing machinery in cells can recognize.
Training data
Splam: Was trained using a high-quality dataset of human donor and acceptor sites. Check out the data curation section.
SpliceAI: Was trained with canonical transcripts only, and does not consider alternative splicing.
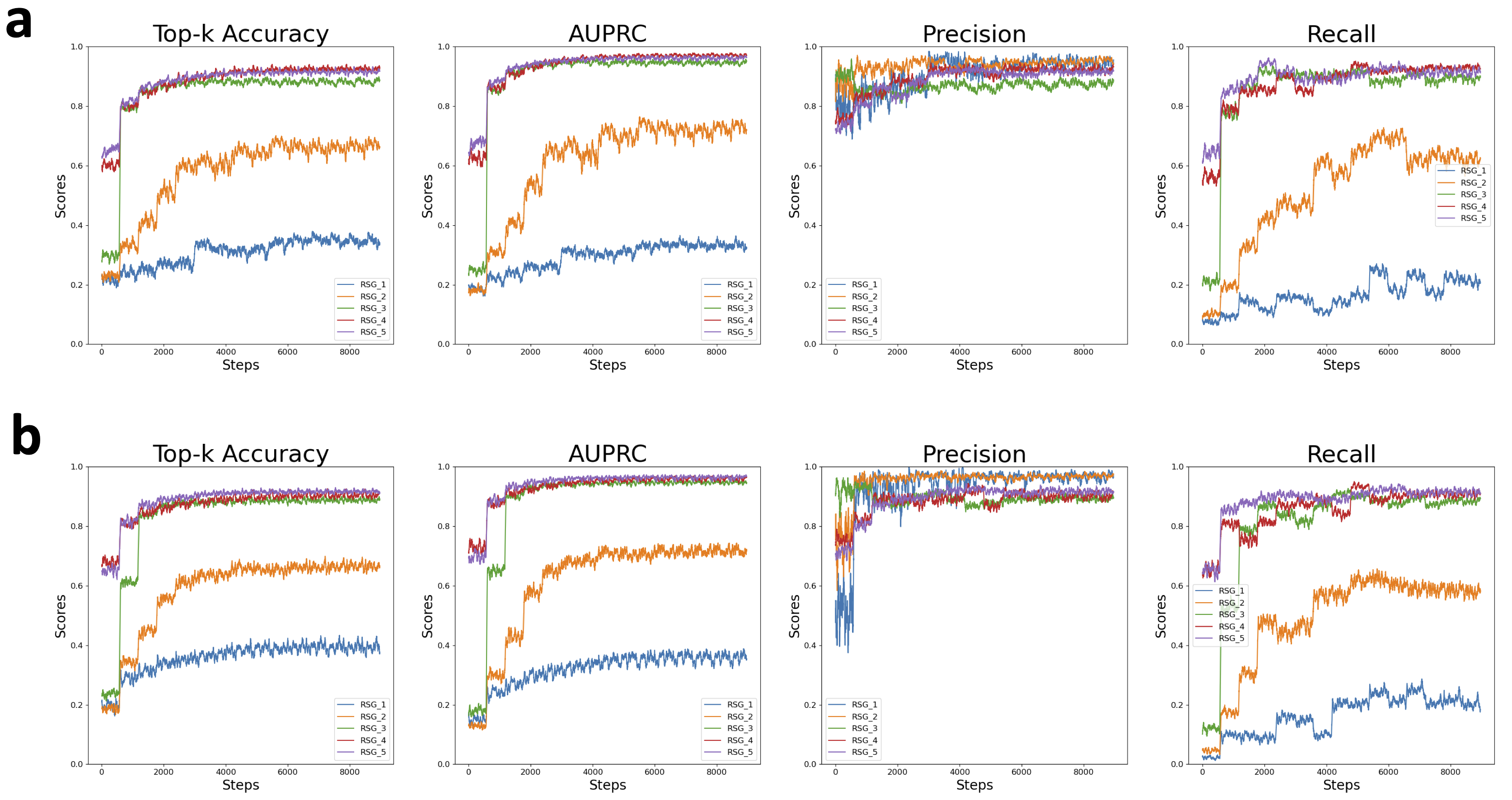
Q: For Splam model design, why do we use five residual groups?
In the design of the Splam model, the inclusion of five residual groups is a result of the experiment to determine the optimal structure for splice site identification. This architectural choice was informed by an ablation study that varied the number of residual groups within the model, aiming to balance complexity with performance efficacy.
Our experiment demonstrated that each additional residual group up to the fifth contributed to the best improvement in the model's performance metrics, including top-k accuracy and the Area Under the Precision-Recall Curve (AUPRC).
Thus, the architecture featuring five residual groups was selected for the final Splam model design, providing the most accurate splice site predition.